预约演示
更新于:2026-04-25
Human Erythropoietin(Shandong Fengjin)
促红细胞生成素(山东丰金)
更新于:2026-04-25
概要
基本信息
药物类型 集落刺激因子 |
别名 FJB1801 |
作用方式 激动剂 |
作用机制 EPO receptor激动剂(促红细胞生成素受体激动剂) |
治疗领域 |
在研适应症 |
非在研适应症- |
原研机构 |
在研机构 |
非在研机构- |
权益机构- |
最高研发阶段临床2期 |
首次获批日期- |
最高研发阶段(中国)临床2期 |
特殊审评- |
登录后查看时间轴
关联
100 项与 促红细胞生成素(山东丰金) 相关的临床结果
登录后查看更多信息
100 项与 促红细胞生成素(山东丰金) 相关的转化医学
登录后查看更多信息
100 项与 促红细胞生成素(山东丰金) 相关的专利(医药)
登录后查看更多信息
17
项与 促红细胞生成素(山东丰金) 相关的文献(医药)2015-07-01·Bulletin of emergency and trauma
Effects of Human Erythropoietin on Functional Outcome of Patients with Traumatic Cervical Cord Injury; A Pilot Randomized Clinical Trial.
Article
作者: Ashraf, Mohammad Hossein ; Farrokhi, Majid Reza ; Alibai, Ehsan Ali ; Mohebali, Navideh ; Baghban, Fahim
OBJECTIVE:
To determine the effects of recombinant human erythropoietin (rhEPO) on functional outcome and disability of patients with traumatic cervical spinal cord injury (SCI).
METHODS:
This was a randomized, double blind, placebo controlled clinical trial being performed in Nemazee and Shahid Rajaei hospitals of Shiraz during a 3-year period from 2011 to 2014. A total number of 20 patients with acute traumatic cervical SCI less than 8 hours after injury were included. We excluded those with anatomic cord dissection, penetrating cord injury and significant concomitant injury. Patients were randomly assigned to receive rhEPO in 500IU/mL dosage immediately and 24-hour later (n=11) or placebo (n=9). All the patient received standard regimen of methylprednisolone. Neurological function was assessed on admission, 1, 6 and 12 months after the injury according to the American Spinal Cord Injury Association (ASIA).
RESULTS:
Overall we include a total number of 20 patients. The mean age of the patients was found to be 40.1±9.5 (ranging from 19 to 59) years. There were 18 (90.0%) men and 2 (10.0%) women among the patients. There was no significant difference between two study groups regarding the baseline characteristics. The baseline ASIA score was comparable between two study groups. The motor and sensory ASIA scores were comparable between two study groups after 1, 6 and 12 months follow-ups. We also found that there was no significant difference between two study groups regarding the motor and sensory outcome in complete cord injury and incomplete cord injury subgroups.
CONCLUSION:
Administration of rhEPO does not improve the functional outcome of patients with traumatic cervical SCI.
2011-11-01·Journal of acquired immune deficiency syndromes (1999)3区 · 医学
Randomized Trial Comparing Dose Reduction and Growth Factor Supplementation for Management of Hematological Side Effects in HIV/Hepatitis C Virus Patients Receiving Pegylated-Interferon and Ribavirin
3区 · 医学
Article
作者: Johnston, Barbara ; Glesby, Marshall J. ; Doonquah, Leleka ; Pearce, Daniel ; Dimova, Rositsa ; Jacobson, Ira M. ; Dove, Lorna ; Aboulafia, David ; Zeremski, Marija ; Aberg, Judy A. ; Talal, Andrew H. ; Rodriguez, Jorge ; Bonilla, Hector ; Liu, Ruei-Chi ; Galpin, Jeffrey ; Hassanein, Tarek
BACKGROUND:
Pegylated-interferon (PEG-IFN) and ribavirin (RBV), current standard treatment for hepatitis C virus (HCV) infection, are frequently associated with neutropenia and anemia, leading to high treatment discontinuation rates in HIV/HCV-coinfected patients. Our objective was to compare the effectiveness of intervening with hematologic growth factors versus dose reductions of standard HCV therapy for the management of treatment-induced hematologic disorders.
METHODS:
Ninety-two HIV/HCV-coinfected, therapy-naive subjects received PEG-IFN alfa-2b 1.5 μg·kg⁻¹·wk⁻¹ and RBV 13 ± 2 mg·kg⁻¹·d⁻¹ for up to 48 weeks. Before treatment initiation, subjects were randomized to subsequently receive growth factors, recombinant human erythropoietin (rHuEPO) and/or granulocyte colony-stimulating factor, or dose reduction (RBV and/or PEG-IFN) for anemia and neutropenia management, respectively. We analyzed the ability of each management strategy to control anemia and neutropenia and the percentage of subjects who achieved a successful treatment outcome according to the different management strategies.
RESULTS:
During treatment, 43 subjects developed anemia (human erythropoietin, n = 24; dose reduction, n = 19), whereas 25 subjects developed neutropenia (granulocyte colony-stimulating factor, n = 10; dose reduction, n = 15). After the intervention, the increase in both hemoglobin and absolute neutrophil counts did not differ between the 2 side effect management strategies. Sustained response percentages were similar comparing anemic and neutropenic subjects regardless of management strategy (anemia: recombinant human erythropoietin, 29% versus dose reduction, 21%, P = 0.92; neutropenia: granulocyte colony-stimulating factor, 40% versus dose reduction, 20%, P = 0.46).
CONCLUSIONS:
Growth factor supplementation and dose reduction do not seem to differ as management strategies for anemia and neutropenia in HIV/HCV-coinfected individuals treated with PEG-IFN/RBV.
2004-01-01·Onkologie
Oncological Management of Pediatric Cancer Patients Belonging to Jehovah’s Witnesses: A Two-Institutional Experience Report
Article
作者: C. Hasan ; U. Bode ; R. Wessalowski ; U. Göbel ; G. Janßen ; C.M. Kramm ; H.-J. Laws ; T. Tenenbaum
OBJECTIVES:
Aim of this study was to analyze the feasibility of oncological treatment in pediatric patients belonging to Jehovah's Witnesses and to describe the changing policy in performing transfusions and supportive care measures at two German pediatric cancer institutions.
PATIENTS AND METHODS:
Over a period of 16 years 21 treatments according to the current cooperative protocols were performed in 14 children of Jehovah's Witnesses. Various hematological supportive care measures such as supplementation with iron, human erythropoietin, interleukin 11, granulocyte colony-stimulating factor and autologous or allogeneic stem cell rescue had been applied. For comparison matched pairs treated in our hospitals not belonging to Jehovah's Witnesses and 50 pediatric and adult oncological patients belonging to Jehovah's Witnesses reviewed from the international literature were analyzed with respect to transfusions and outcome.
RESULTS:
So far, 9 of 14 children are surviving 16-195 months (median 26 months). During the primary therapy they received markedly less transfusions than the control cohort (-39,1% red blood cell transfusions and -37,5% platelet transfusions). The review of 50 reported cases showed that oncological therapy can also be successfully performed with a restricted transfusion regimen in children and particularly in adults.
CONCLUSION:
Pediatric cancer patients belonging to Jehovah's Witnesses can be treated similarly to other patients. A restrictive transfusion policy and the broad application of hematopoietic supportive care measures may reduce transfusions. This treatment policy and a continuous collaboration with the Hospital Liaison Committee for Jehovah's Witnesses appears to create an oncological treatment situation with a high compliance of patients and parents where court orders may not be necessary.
2
项与 促红细胞生成素(山东丰金) 相关的新闻(医药)2026-04-22
在科研实践中,研究者很容易陷入“技术舒适区”——习惯用已掌握的技术去框定研究问题。然而,HIF信号机制的探究过程,恰恰是对这种路径依赖的突破:科学问题应当定义技术路径的选择,而非被熟悉的工具所局限。
最具颠覆性的科学问题,往往隐藏在人们习以为常,甚至视为理所当然的生物学现象背后。
氧气,是几乎所有真核生物能量代谢的基石,更是生命存续不可或缺的核心物质。从海拔数千米的高原攀登时的低氧环境,到胚胎发育过程中血管新生的动态变化,再到实体肿瘤内部氧分压极低的微环境,我们体内的每一个细胞,都无时无刻不在面对外界氧浓度的波动与挑战。
为了维持正常的生存与生理运作,细胞必须精准感知氧气水平的变化,进而启动一系列适应性反应。而细胞这种精准的氧感知与适应能力,背后隐藏着一套复杂而精妙的分子机制,这一机制的揭晓,离不开三位科学家的不懈探索。
2019年10月7日,诺贝尔生理学或医学奖授予威廉·凯林(William G. Kaelin Jr.)、彼得·拉特克利夫(Sir Peter J. Ratcliffe)和格雷格·塞门扎(Gregg L. Semenza),以表彰他们发现了“细胞感知和适应氧气供应”的分子机制。
这一发现历经近三十年,他们的工作从关于贫血与造血的问题出发,揭示了一个困扰科学界数十年的核心谜题:细胞究竟如何“知道”自己处于缺氧状态,并启动一系列精确的适应性反应。他们的研究不仅在基础科学领域揭示了生命活动中关键的适应性过程,更在临床医学领域为贫血、心血管疾病以及恶性肿瘤等多种疾病提供了全新的治疗靶点与干预策略。
图1. 2019年诺贝尔生理学或医学奖的三位得主:威廉·凯林(William G. Kaelin Jr.)、彼得·拉特克利夫(Sir Peter J. Ratcliffe)和格雷格·塞门扎(Gregg L. Semenza)。图片来源:诺贝尔奖官网
1774年,英国化学家约瑟夫・普利斯特里首次分离出氧气;随后,安托万・拉瓦锡通过经典的汞燃烧实验以及豚鼠的呼吸实验,提出动物的呼吸作用实质上是缓慢的氧化过程,揭示了“燃烧”与“生物呼吸”的共性。自此,人类开始明白,生命的存续,本质上是一个持续利用氧气完成能量代谢的过程。
但在此后的近200年里,科学界对缺氧适应的认知,始终停留在器官与系统层面。19世纪末,生理学家观察到一个明确的现象:人类或动物进入高原低氧环境后,外周血中的红细胞数量会显著增加,以此提升血液的携氧能力,适应低氧环境。1906年,法国科学家Carnot和Deflandre在实验中首次提出,动物体内存在一种可随血液循环的“促红细胞生成因子”,正是这种物质介导了缺氧后的红细胞增多。这一因子,就是后来被我们熟知的促红细胞生成素(EPO)。
图2. 早期关于高海拔缺氧适应的生理学观察实验,展示了进入低氧环境后红细胞与血液携氧能力的代偿性变化。图片来源:诺贝尔奖官网
这一发现开启了缺氧生理研究的全新阶段,但也带来了新的谜题:究竟是什么信号,让肾脏感知到身体的缺氧状态,并启动EPO的合成与分泌?
在随后的大半个世纪里,科学家们逐步厘清了EPO的生理功能:它由肾脏皮质的管周成纤维细胞合成,作用于骨髓红系祖细胞,促进其增殖分化,最终提升红细胞数量。1977年,科学家从再生障碍性贫血患者的尿液中成功纯化出EPO蛋白;1985年,EPO基因被成功克隆,重组人EPO也随即问世,成为肾性贫血治疗的里程碑药物。
但核心的科学问题依然没有答案:缺氧信号,究竟如何调控EPO基因的表达?
彼时,主流观点认为,哺乳动物的缺氧感知主要依赖颈动脉体与主动脉体的外周化学感受器,通过神经反射调节呼吸与循环功能;而单个细胞对氧气的自主感知,仅被视作次要的、被动的效应。没人能料到,正是这一被主流忽视的效应,才是氧感知机制的核心,更隐藏着一个几乎适用于所有哺乳动物细胞、高度保守的基因调控网络。
而率先在这一研究领域取得突破的,正是后来的诺贝尔奖得主——Gregg L. Semenza。
破局的关键:从EPO调控元件,到HIF的发现
Semenza的研究起点,正是那个困扰学界数十年的问题:EPO基因的表达,为何能被缺氧精准调控?
早在二十世纪70年代,科学界便已明确EPO的产生与分泌具备三个特征:其一,高度的组织特异性;其二,严格的发育阶段特异性(胚胎期主要由肝脏产生,成年后则主要由肾脏分泌);其三,极强的诱导性,即在面临缺氧、贫血或氯化钴暴露时,体内EPO的表达水平会呈现出爆发式的增长。正是这种与氧气浓度紧密偶联的诱导表达特性,使得EPO基因成为破译氧气感知之谜的理想分子模型。
在EPO基因被克隆后,Semenza敏锐地意识到,要找到缺氧调控的分子机制,关键是先找到EPO基因上负责响应缺氧信号的顺式作用元件。这是一个极具针对性的研究思路:如果把EPO基因的表达比作一盏灯,那么顺式作用元件就是开关,而我们首先要找到这一开关在基因序列上的具体位置。
为此,Semenza团队开展了一系列精细的报告基因实验:他们将人EPO基因的不同片段与报告基因融合,构建了一系列突变载体,转染到肝癌细胞系Hep3B中,对比常氧与缺氧条件下的报告基因活性。
经过反复的序列截短与突变验证,Semenza团队于1992年取得了首个里程碑式发现:在EPO基因的3’端非编码区,存在一段长度为50bp的DNA序列,它是缺氧诱导EPO表达的必需元件——该序列存在时,EPO基因可被低氧条件诱导表达;一旦删除,缺氧诱导效应便完全消失。他们将这段序列命名为缺氧响应元件(Hypoxia Response Element, HRE)。后续研究中,Semenza进一步鉴定出HRE内18bp的核心序列是发挥作用的关键区域。
图3. 利用人EPO基因不同片段开展的转基因小鼠实验,寻找负责响应缺氧信号的顺式作用元件的位置。(Semenza et al., 1990)
图4. 人EPO基因缺氧响应元件(HRE)的功能分析,通过扫描突变验证了核心序列在缺氧诱导表达中的关键作用。(Semenza and Wang, 1992)
HRE序列的鉴定,意味着科学家终于有了明确的“诱饵”,去捕捉那个在缺氧条件下结合HRE、启动基因表达的关键转录因子。为了证明缺氧细胞内确实存在一种能结合在EPO基因上的蛋白,Semenza提取了常氧和缺氧细胞的核蛋白,并将其与带有放射性同位素标记的HRE序列DNA探针混合孵育。在电泳跑胶时,缺氧组的探针因为结合了巨大的蛋白复合体,移动速度变慢,出现了明显的滞后条带。
图5. 电泳凝胶阻滞实验结果,显示缺氧细胞的核蛋白与HRE序列探针结合后,形成了明显的滞后条带,证实了结合蛋白的存在。(Semenza and Wang, 1992)
接下来,克隆结合该序列的转录因子似乎只需一步之遥。最开始,Semenza希望利用噬菌体cDNA表达文库进行Southwestern blotting筛选。这套系统曾成功钓出过众多转录因子——只需用放射性同位素标记的HRE双链DNA作为探针,去孵育表达了蛋白的硝酸纤维素膜,发黑的斑点即代表目标基因。然而,这一经典技术在寻找缺氧诱导因子时,遭遇了彻底的失败。数以百万计的克隆中,没有一个能与探针稳定结合。
但是Semenza并未放弃,转而采用生化方法纯化并鉴定目标蛋白。这是一项极具挑战性的工作:在当时的技术条件下,从细胞核提取物中分离出序列特异性转录因子,难度堪比大海捞针——这类蛋白在细胞内的表达丰度极低,且仅在缺氧条件下才会激活并结合DNA,稳定性极差。更棘手的是,Semenza的实验室当时专注于分子遗传学研究,甚至连最基础的组分收集器都未配备。
由于没有捷径可循,Semenza团队只能通过大规模细胞培养,借助DNA亲和层析技术,逐步富集并纯化能特异性结合HRE的蛋白。经过数年的筛选与纯化,他们终于成功克隆并鉴定出这种在缺氧条件下结合HRE、启动靶基因转录的转录因子,将其命名为缺氧诱导因子1(Hypoxia-Inducible Factor 1, HIF-1)。
图6. 利用DNA亲和层析技术,从细胞核提取物中逐步富集并纯化出缺氧诱导因子1(HIF-1)。(Wang et al., 1995)
更关键的是,他们发现HIF-1并非单一蛋白,而是由两个亚基组成的异二聚体:
一个是组成型表达、不受氧浓度调控的HIF-1β(也被称为ARNT),在常氧与缺氧条件下均稳定存在;
另一个是HIF-1α,它的蛋白稳定性与转录活性完全由氧浓度精准调控:在常氧条件下,HIF-1α几乎无法被检测到,蛋白会被迅速降解;而在缺氧条件下,HIF-1α快速稳定积累,进入细胞核与HIF-1β形成二聚体,结合靶基因的 HRE序列,启动转录。
图7. HIF-1蛋白异二聚体(HIF-1α与HIF-1β)的纯化结果及其关键结构域示意图。(Wang et al., 1995)
与此同时,Peter J. Ratcliffe团队的研究,进一步拓展了这一发现的边界。他们通过实验证实,HIF介导的缺氧响应,并非产生EPO的细胞特有的调控机制,而是一个普适于几乎所有哺乳动物细胞的核心通路:无论是血管内皮细胞、心肌细胞,还是肿瘤细胞,都存在HIF-1α的氧依赖调控,且HRE元件广泛存在于数十种缺氧响应基因的启动子或增强子区域,包括血管内皮生长因子(VEGF)、葡萄糖转运体(GLUT1)、糖酵解关键酶等。
图8. 多细胞系验证实验,证实HIF-1α的氧依赖调控广泛存在于肝癌细胞、单核细胞、成纤维细胞等多种哺乳动物细胞系中。(Maxwell et al., 1993)
图9. 探针结合实验,证明HIF复合体可以结合包括EPO、葡萄糖转运体和糖酵解关键酶在内的多种靶基因的HRE元件。(Semenza et al., 1994)
这意味着,科学家们找到的并非仅仅是EPO的调控开关,而是一套细胞应对缺氧的通用机制。这套程序,同时调控着红细胞生成、血管新生、代谢重编程、细胞增殖与凋亡等一系列关键生理过程,是细胞适应低氧环境的核心指挥系统。
至此,细胞应对缺氧的适应效应已逐渐清晰,但一个更核心的谜题仍悬而未决:氧分子究竟如何精准调控HIF-1α的蛋白稳定性?常氧条件下,HIF-1α为何会被迅速降解?这一过程又如何被氧浓度精准控制?
解答这个谜题的关键线索,意外地来自一种罕见的遗传性肿瘤综合征,而解开这个谜题的,正是William G. Kaelin Jr.与Peter J. Ratcliffe的团队。
机制的闭环:VHL蛋白、羟化修饰与氧感知的分子开关
故事的另一条主线,始于一种名为希佩尔-林道综合征(Von Hippel-Lindau Syndrome, VHL综合征)的罕见遗传病。这是一种常染色体显性遗传疾病,患者因VHL基因的突变,在生命过程中极易发生双等位基因失活,进而引发多发的血管母细胞瘤、肾透明细胞癌、嗜铬细胞瘤等肿瘤。这些肿瘤有一个极其显著的特征:高度血管化,即使在常氧环境下,也会持续大量分泌VEGF、EPO等缺氧响应基因,表现出“假性缺氧”的表型,仿佛细胞始终处于缺氧环境中。
William G. Kaelin Jr.是VHL综合征研究领域的顶尖专家。VHL综合征的病理特征使他敏锐地意识到,VHL基因的功能缺失,与细胞的缺氧响应通路之间,必然存在着直接的因果关联。而VHL基因,很可能就是解开HIF-1α氧依赖降解之谜的关键。
1999年,Kaelin团队与Ratcliffe团队几乎同时发表了关键研究成果,共同揭示了VHL蛋白的核心功能:作为E3泛素连接酶复合物的核心组分,VHL蛋白可直接结合HIF-1α,介导其泛素化修饰,进而使其被蛋白酶体识别并降解。
他们的实验结果显示:
在VHL基因缺失的肾癌细胞中,即便处于常氧条件,HIF-1α仍能稳定积累,并持续激活下游缺氧响应基因;
向这些细胞中回补野生型VHL基因后,细胞在常氧下快速降解HIF-1α的能力完全恢复,“假性缺氧”表型也被逆转;
突变体实验进一步证实,VHL蛋白与HIF-1α的直接结合,是HIF-1α发生泛素化降解的必要前提。
图10. VHL基因调控HIF-α的实验,显示在VHL缺陷细胞中回补野生型VHL基因后,细胞在常氧下降解HIF-α的能力得以恢复。(Maxwell et al., 1999)
这一发现,完美解释了VHL综合征患者的肿瘤表型:VHL基因失活后,HIF-1α在常氧下也无法被降解,持续激活血管新生、糖酵解等通路,最终驱动肿瘤的发生发展。
但同时,一个新的核心问题再次出现:VHL蛋白只会在常氧下结合HIF-1α,在缺氧条件下则完全不结合,这种氧依赖的结合能力,究竟是如何实现的?
答案的揭晓,来自2001年发表在《Science》上的三篇背靠背论文,Kaelin团队、Ratcliffe团队与Pugh团队,几乎同时破解了这个氧感知的核心谜题。
他们发现,HIF-1α蛋白的氧依赖降解,完全依赖于其特定氨基酸残基的翻译后修饰——在HIF-1α的氧依赖降解结构域(ODD)中,存在两个高度保守的脯氨酸残基(Pro402和Pro564),在常氧条件下,这两个脯氨酸会被一类名为脯氨酸羟化酶(Prolyl Hydroxylase Domain, PHD)的酶催化,发生4-羟化修饰;而缺氧条件下,这个羟化反应则会完全停止。
更关键的是,羟化修饰后的HIF-1α,是VHL蛋白识别并结合的底物。VHL蛋白的β结构域,能精准识别羟化后的脯氨酸残基,形成高亲和力的结合;而未发生羟化的HIF-1α,完全无法被VHL蛋白识别,自然也就不会发生泛素化降解。
图11. HIF-1与HIF-2蛋白的结构域,重点标注了氧依赖降解结构域(ODDD)上发生关键羟化修饰的脯氨酸残基(P402和P564)。图片来源:诺贝尔奖官网
至此,细胞氧感知的完整分子机制,终于形成了完美的闭环,而氧分子本身,正是这个闭环中最核心的分子开关:
常氧状态下:氧分子作为PHD酶的必需底物,维持着PHD的催化活性;PHD 持续催化HIF-1α的脯氨酸羟化修饰,羟化后的HIF-1α被VHL-E3泛素连接酶复合物识别,发生多聚泛素化,最终被蛋白酶体快速降解,细胞内几乎检测不到HIF-1α蛋白。
缺氧状态下:氧分压下降导致PHD酶的催化活性迅速丧失,HIF-1α的羟化修饰停止;未被羟化的HIF-1α无法被VHL蛋白识别,因此在细胞内快速稳定积累,进入细胞核与HIF-1β形成异二聚体,结合靶基因启动子/增强子区域的HRE元件,启动数百种缺氧响应基因的转录,完成细胞的缺氧适应重编程。
图12. 细胞氧感知核心机制(PHD-HIF-VHL轴)闭环示意图,展示了常氧下的HIF-α羟化降解过程与缺氧下的入核转录过程。(Sugahara et al., 2017)
后续的研究进一步完善了这一机制。FIH(Factor Inhibiting HIF,即HIF抑制因子)作为另一类依赖氧气和2-酮戊二酸的双加氧酶,构成了氧感知通路中独立于蛋白丰度调控的第二重精细调控机制——转录活性门控。
常氧状态下:FIH同样以氧分子为催化底物,特异性地对HIF-1α的C端反式激活结构域(C-TAD)上的一个高度保守的天冬酰胺残基进行羟基化修饰。这一化学加合物产生了显著的空间位阻,直接阻断了HIF-1α与关键转录共激活因子(如CBP/p300组蛋白乙酰转移酶)的结合。因此,即使有极少量的HIF-1α逃避了pVHL的降解进入细胞核,也无法启动下游基因表达。
缺氧状态下:随着氧分压的降低,FIH的催化活性受到抑制,天冬酰胺的羟基化修饰停止。未被修饰的C-TAD恢复了与CBP/p300的高亲和力结合,使得HIF复合体恢复转录活性。
从EPO的表型发现,到HRE元件的鉴定,从HIF-1的克隆纯化,到VHL蛋白功能的揭示,最终到脯氨酸羟化修饰这个氧感知核心开关的发现,三个独立的研究团队,从不同的研究起点出发,跨越十余年的时间,共同完成了这幅细胞氧感知机制的完整拼图。这一整套高度保守、精准调控的分子通路,也被学界命名为PHD-HIF-VHL轴,成为现代分子生理学最重要的基石之一。
从基础机制到临床转化:氧感知研究的医学价值
从进化生物学的视角来看,HIF介导的氧气感知机制可追溯至约八亿年前,其出现时间与地球大气氧含量达到巴斯德点(约为当前大气氧水平的百分之一)的节点高度契合,为寒武纪生命大爆发奠定了坚实的生物能量学基础。
而在今天,这一古老机制仍在不断焕发新的生机。HIF氧感知机制的阐明,不仅解答了生命科学领域最基础的生理问题,更彻底改变了我们对多种疾病病理机制的认知,为临床治疗带来了颠覆性突破。
肾性贫血的治疗突破
肾性贫血是慢性肾脏病(CKD)最常见的并发症,其核心病因是肾脏EPO分泌不足,导致红细胞生成障碍。在HIF机制被阐明之前,肾性贫血的治疗只能依赖外源性重组人EPO注射,不仅需要频繁给药,还存在高血压、血栓等不良反应风险。
而PHD-HIF轴的发现,带来了全新的治疗策略:通过抑制PHD酶的活性,稳定HIF蛋白,从而生理性地促进内源性EPO的分泌,同时上调铁代谢相关基因,改善铁利用障碍。基于这一机制研发的低氧诱导因子脯氨酸羟化酶抑制剂(HIF-PHI),已经成为肾性贫血治疗的一线药物。目前,全球已有多款HIF-PHI药物获批上市,包括罗沙司他、达普司他、伐度司他等,数百万慢性肾脏病患者因此受益。这也是诺奖成果从基础研究快速转化为临床药物的经典范例。
肿瘤治疗的全新方向
在绝大多数实体肿瘤中,肿瘤微环境都存在显著的缺氧特征,而HIF通路的持续激活,是肿瘤细胞适应缺氧微环境的核心机制:HIF能驱动肿瘤细胞的糖酵解重编程(瓦博格效应)、血管新生、侵袭转移、免疫逃逸,以及化疗耐药。
而基于HIF通路的肿瘤治疗策略,已经成为全球抗肿瘤药物研发的热门方向:针对HIF-2α的特异性抑制剂,已经在VHL相关肾透明细胞癌的治疗中取得了突破性的临床疗效,成为晚期肾癌的二线治疗方案;靶向PHD、VHL的药物研发,以及HIF通路与免疫检查点抑制剂的联合治疗方案,正在全球范围内开展大量临床试验,有望为更多实体瘤患者带来生存获益。
除此之外,HIF氧感知机制还在胚胎发育、高原适应、肺部疾病、代谢性疾病、炎症免疫等领域,展现出了极其重要的生理与病理意义。可以说,几乎所有与缺氧相关的生理病理过程,都离不开PHD-HIF-VHL轴的调控,而这一机制的发现,为整个生命科学与医学领域,打开了一扇全新的大门。
图13. 基于细胞氧感知机制研发的临床转化药物,包括用于治疗肾性贫血的HIF-PHI药物(如罗沙司他)和靶向HIF通路的抗肿瘤药物。图片来源于网络。
诺奖背后的科研启示
回顾这三位科学家跨越近三十年的探索历程,除了精妙的分子机制本身,这段科学史更像是一面镜子,反映出了生命科学研究中最核心的几项底层逻辑。结合他们在研究过程中的关键选择,我们或许可以提炼出以下三点深刻的科研启示:
一、以科学问题为导向的“技术勇气”(Technical Courage)
在科研实践中,研究者很容易陷入“技术舒适区”——习惯用已掌握的技术去框定研究问题。然而,HIF信号机制的探究过程,恰恰是对这种路径依赖的突破:科学问题应当定义技术路径的选择,而非被熟悉的工具所局限。
当Gregg Semenza面对噬菌体表达文库筛选的彻底失败时,他果断放弃了当时最主流、自己也最熟悉的遗传学与分子克隆捷径。为了拿到那个未知的异源二聚体,他毅然转向了极其繁琐的大规模生化纯化。
同样,Peter Ratcliffe在探究氧感知修饰酶的过程中,最初的假设是某种磷酸化激酶。但当磷酸化假设被全面推翻后,他并没有在原有的认知框架内打转,而是迅速跨界转向了酶学、底物序列预测,甚至引入了线虫遗传筛选模型,最终精准锁定了PHD家族。
值得注意的是,Semenza和Ratcliffe两人都是在求学生涯较晚的阶段才深入接触分子生物学与生物化学,但这并未成为他们的技术壁垒。在如今单细胞测序、冷冻电镜等高通量/高分辨技术高度发达的今天,我们或许更应该反问自己:在现有的技术条件下,我们是否有勇气为了那个“必须回答的问题”,去跨越自己知识与能力的边界?
二、科学突破是跨领域的“集体事业”
HIF-VHL-PHD机制的最终揭示,完美展现了科学界开放交流与协同互补的巨大力量:
1993年,在马萨诸塞州Woods Hole举行的一次学术会议上,Semenza和Ratcliffe首次深入交流了各自的研究。一个是拿着HIF序列寻找上游,一个是在证明氧感知具有泛组织普遍性。这次早期的碰面,开启了整个领域更为深入的协作探索。
1998年,在VHL家族联盟年会上,Ratcliffe实验室的博士后与William Kaelin进行了关键的数据交换。他们分享了关于HIF-2α在VHL缺陷细胞中异常积累的未发表信息,这一无私的学术共享,直接打通了这两个原本平行的研究轨道,极大地加速了机制的最终揭示。
现代生命科学早已告别了单打独斗的时代,每一个团队都在解决一个局部微小的问题,但正是这些看似不起眼的进步,最终汇聚成了生命科学史上的浓墨重彩。
三、基础研究的问题往往隐藏在“显而易见”的生理现象中
最具颠覆性的科学问题,往往隐藏在人们习以为常,甚至视为理所当然的生物学现象背后。人体在高海拔缺氧时会通过代偿增加红细胞,这是每个医学生都熟知的生理现象。然而,三位科学家没有止步于描述这种代偿,而是敏锐地追问:那个能感知到氧气浓度并决定开启EPO转录的分子究竟是什么?
这也提醒我们,实验室最值得回答的问题,不只存在于最前沿的想象中,也同时存在于那些被写进教科书、却从未被彻底解释的生理活动里。
参考文献
Semenza, G. L. & Wang, G. L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12, 5447-5454 (1992).
Maxwell, P. H., Pugh, C. W. & Ratcliffe, P. J. Inducible operation of the erythropoietin 3' enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism. Proc Natl Acad Sci U S A 90, 2423-2427 (1993).
Semenza, G. L., Roth, P. H., Fang, H. M. & Wang, G. L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269, 23757-23763 (1994).
Wang, G. L. & Semenza, G. L. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 270, 1230-1237 (1995).
Forsythe, J. A. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16, 4604-4613 (1996).
Iliopoulos, O., Levy, A. P., Jiang, C., Kaelin, W. G., Jr. & Goldberg, M. A. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A 93, 10595-10599 (1996).
Maxwell, P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271-275 (1999).
Epstein, A. C. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43-54 (2001).
Ivan, M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464-468 (2001).
Mahon, P. C., Hirota, K. & Semenza, G. L. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 15, 2675-2686 (2001).
2026-01-26
·药时空
摘要:从1921年胰岛素的首次分离,到如今琳琅满目的单克隆抗体、双特异性抗体乃至抗体偶联药物(ADC),生物制剂已经彻底改变了现代医学的格局。它们不再是简单的天然提取物,而是经过精密设计的“生物导弹”,在治疗癌症、自身免疫病、代谢性疾病等方面大放异彩。这篇文章,我们将一起回顾这场激动人心的“进化史”,看看科学家们如何通过蛋白工程、Fc工程等技术,不断优化这些“大分子药物”,让它们变得更精准、更持久、更安全。
第一部分:从“天然”到“重组”,小蛋白的大革命
一切始于胰岛素。这个由51个氨基酸组成的小蛋白,最初从动物胰腺中提取,不仅产量有限,还容易引起免疫反应。直到重组DNA技术的出现,才实现了人胰岛素的大规模、高质量生产,这是生物制药史上的第一座里程碑。你看,技术的突破直接决定了药物的可及性。
图1:胰岛素的进化之路
类似的成功故事还有促红细胞生成素(Epogen)和粒细胞集落刺激因子(Neupogen),它们分别用于治疗贫血和中性粒细胞减少症。但这些小分子量蛋白有个通病:在体内半衰期太短,容易被肾脏快速清除,患者需要频繁注射,非常不便。
怎么办?科学家们想出了“嫁接”的妙招。比如,把GLP-1(一种降糖激素)与人血清白蛋白或抗体Fc片段融合,就诞生了阿必鲁肽和度拉糖肽这类一周只需注射一次的新药,大大提升了患者依从性。这背后的原理,我们后面会详细说。
第二部分:抗体的“人源化”战争
如果说小蛋白是先锋,那么抗体就是生物制药军团的主力部队。但早期直接用鼠源抗体给人治病,身体会把它当“外敌”攻击,产生抗药物抗体(ADA),导致药效降低甚至副作用。
于是,一场降低免疫原性的“人源化”战争打响了。第一步是制造嵌合抗体,比如英夫利西单抗和利妥昔单抗,它们把老鼠的“识别头”(可变区)接在人的“身体”(恒定区)上。这还不够,进一步的技术把老鼠的识别区也尽可能地“伪装”成人的样子,这就是人源化抗体,像曲妥珠单抗(赫赛汀)就是代表。
图2:治疗性单克隆抗体的各种“版本”
终极目标是全人源抗体。现在我们可以用噬菌体展示等技术,直接从人类基因库里筛选出完全属于人的抗体序列,或者培育出携带人类抗体基因的转基因小鼠来生产抗体。纳武利尤单抗(O药)就是这类技术的结晶。你看,从“像人”到“是人”,每一步都为了更安全。
第三部分:抗体的“心脏”:Fc区域的奥秘
抗体长得像个“Y”,上半部分两个叉负责识别目标(抗原),下半部分的“树干”就是Fc区域。可别小看它,这简直是抗体的“智能心脏”。
首先,Fc决定了抗体的“性格”。IgG1擅长调动免疫细胞去杀伤靶细胞(比如癌细胞),这叫抗体依赖性细胞介导的细胞毒性作用(ADCC);而IgG4则比较“温和”,适合用于只需要阻断信号、不需要引起杀伤的场合。工程师们现在还能通过定点突变,随意增强或消除这些功能,定制我们想要的抗体。
图3:Fcγ受体在免疫细胞上的表达及其功能
更神奇的是,Fc还是抗体的“长寿秘诀”。它能与体内的新生儿Fc受体(FcRn)结合。这个受体像是个“回收站”,在血液中保护抗体不被降解,从而将抗体的半衰期延长到数周之久。前面提到的白蛋白融合技术,也是利用了白蛋白也能被FcRn回收的特性。理解了Fc,你就掌握了设计长效生物制剂的核心密码。
第四部分:下一代“生物导弹”:更复杂,更精准
有了抗体这个平台,科学家们的想象力彻底放飞了。他们开始设计更复杂的“组合型”药物,我管它们叫“超级生物导弹”。
双特异性抗体是典型代表,它一个抗体能同时结合两个靶点。比如贝林妥欧单抗,一头拉着癌细胞(CD19),一头拉着免疫T细胞(CD3),强行让T细胞精准杀伤肿瘤,威力巨大。现在,这种思路甚至被拓展到自身免疫病的治疗中,前景广阔。
抗体偶联药物(ADC)则像是“精准化疗”。它用抗体作为导航头,后面挂着一个剧毒的小分子化疗药物(payload)。抗体把毒素直接送到肿瘤细胞内部再释放,实现“定点爆破”,大大减少了传统化疗的全身毒性。恩美曲妥珠单抗和德曲妥珠单抗在乳腺癌治疗上的成功,尤其是后者更优的疗效,直接带动了整个ADC领域的复兴。
还有放射偶联药物,把放射性同位素装在抗体上,进行内照射治疗;以及条件活性抗体,像安装了“保险栓”,只在肿瘤微环境这种特定条件下才被激活,进一步减少副作用。这些创新,都在让治疗变得越来越“聪明”。
第五部分:光环之下,挑战犹存
当然,生物制剂并非完美无瑕。免疫原性风险虽然降低但从未消失,抗药物抗体(ADA)仍是需要密切监测的问题。有些疗法,尤其是强力激活免疫系统的,可能引发凶险的细胞因子释放综合征(CRS),需要医生有丰富的处理经验。
此外,脱靶效应、长期安全性数据的积累、高昂的生产成本,都是摆在科学家和药企面前的现实挑战。每一款新药上市的灿烂光环背后,都是无数次的优化、失败和严谨的验证。
结语:
回顾生物制剂的发展,就是一部从“发现自然”到“设计生命”的微观史诗。早期的药物是“找到什么用什么”,而现在,我们进入了“需要什么就设计什么”的时代。蛋白工程、人工智能辅助设计、新型偶联技术,这些工具让我们有能力针对疾病的复杂机制,定制出多功能的治疗模块。
未来的生物制药,比拼的不仅仅是发现一个靶点,更是工程化设计的能力。如何让药物更精准地抵达病灶、更持久地发挥作用、更聪明地响应人体信号,将是研发的核心。这场关于生命的精密设计竞赛,才刚刚进入最精彩的章节,而最终的受益者,将是每一位患者。
识别微信二维码,可添加药时空小编
请注明:姓名+研究方向!
抗体药物偶联物引进/卖出生物类似药
100 项与 促红细胞生成素(山东丰金) 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 血液疾病 | 临床2期 | 中国 | 2022-01-30 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
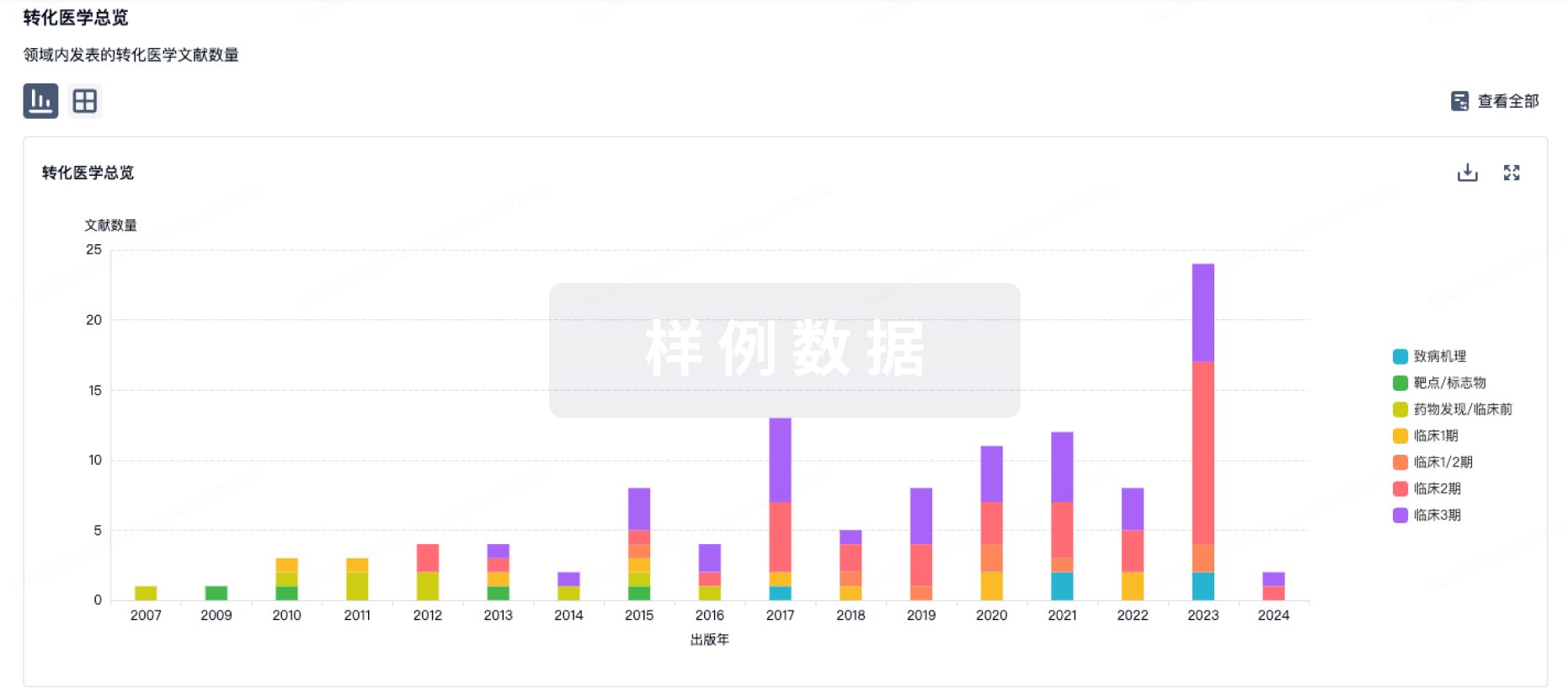
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
生物类似药
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用