预约演示
更新于:2026-01-12
SCN9A Candidate
更新于:2026-01-12
概要
基本信息
原研机构 |
在研机构- |
非在研机构 |
权益机构- |
最高研发阶段无进展早期临床1期 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |

登录后查看时间轴
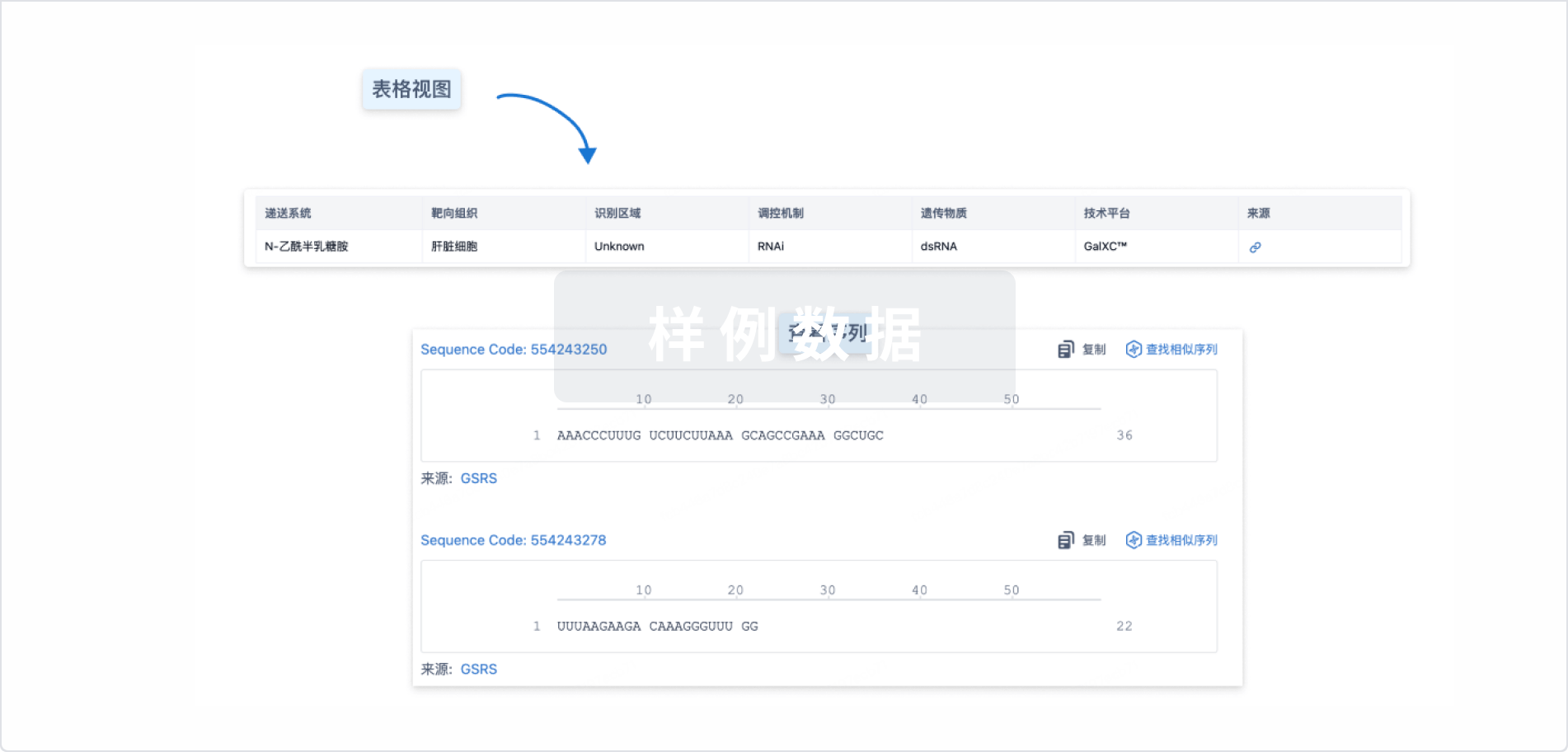
结构/序列
使用我们的RNA技术数据为新药研发加速。
登录
或
关联
1
项与 SCN9A Candidate 相关的临床试验ChiCTR-RCH-10000970
SCN9A gene heterozygous mutation may cause a partial deletion of the feeling of pain
开始日期2008-10-01 |
申办/合作机构 华中科技大学同济医学院 [+1] |
100 项与 SCN9A Candidate 相关的临床结果
登录后查看更多信息
100 项与 SCN9A Candidate 相关的转化医学
登录后查看更多信息
100 项与 SCN9A Candidate 相关的专利(医药)
登录后查看更多信息
8
项与 SCN9A Candidate 相关的新闻(医药)2025-10-27
全书总结提炼核心观点: 《基因密码》是一部融合了前沿科学、真实医患故事和深刻人文关怀的作品。它以作者的亲身经历为主线,生动记录了过去十余年基因组学从高深莫测的科研领域迅速走向临床应用,并彻底改变疾病诊断、治疗和预防方式的革命性历程。书中强调,基因组不仅是生命的蓝图,更是解开个体健康之谜、实现“先发制人”的精准医疗的关键,但其发展也伴随着复杂的伦理、社会和经济挑战。作者的主要论证过程/叙事结构:1. 早期探索与奠基(第一部分): 通过“零号患者”斯蒂芬·奎克和“分子尸检”里奇·奎克的案例,展示全基因组测序分析的初步尝试和巨大潜力,以及索莱科萨/因美纳等公司如何推动测序技术商业化、普及化。强调了团队合作和参考基因组的重要性。2. 基因诊断的力量(第二部分): 聚焦于未确诊疾病网络(UDN)的工作,通过一系列感人的患者故事(如NGLY1缺乏症、MEPAN综合征等),展现基因组学如何终结患者的“诊断奥德赛”,发现新疾病,并启发治疗思路。强调了临床观察、国际合作与数据共享的价值。3. 心脏相关的基因奥秘(第三部分): 以心脏疾病为切入点,深入讲述长QT综合征、肥厚型心肌病、卡尼综合征等遗传性心脏病的发现史、分子机制以及基因组学在危重症救治(如新生儿快速测序、嵌合体诊断、长读长测序识别结构变异)和推动基础研究(如肌球蛋白功能)中的突破性应用。4. 迈向精准医疗的未来(第四部分): 展望基因组学在更广泛领域的应用,包括通过研究“超人基因”开发新药、利用大规模人群队列(如“我们所有人”项目、英国生物样本库)实现常见病的精准风险预测、基因治疗(包括RNA疗法和CRISPR基因编辑)的黄金时代,以及基因组学在应对全球公共卫生挑战(如COVID-19大流行)中的核心作用。贯穿全书的新奇/重要知识点:技术革命: 从桑格测序到下一代测序(NGS)、长读长测序、纳米孔测序的演进,以及CRISPR基因编辑等颠覆性技术的诞生与应用。诊断突破: “分子尸检”、体细胞嵌合现象的临床诊断、利用iPSC和动物模型进行功能验证、发现新疾病基因和综合征。治疗创新: 药物重用、基于基因信息的个性化用药、基因治疗(基因替代、RNA干扰、基因编辑)的成功案例和“N-of-1”定制疗法。大规模数据驱动: 大型人群队列研究(如UK Biobank, All of Us)、公共数据库(如gnomAD)、多基因风险评分(PRS)对理解复杂疾病和实现精准预防的重要性。伦理与社会: “偶发性基因组”、知情同意、基因隐私、胚胎基因编辑的伦理争议、基因治疗药物的成本与可及性等。科学与人文的交织: 强调患者故事是推动科学进步的动力,医生与科学家的人文关怀、团队协作精神以及对未知领域的不懈探索。历史视角: 书中常穿插对相关疾病发现史、关键技术发明史和重要科学人物的介绍,赋予了内容深度和趣味性。推荐序 (尹传红)核心观点: 《基因密码》是一部真诚且值得细品的医学之书,它揭示了基因组学如何引领我们进入“先发制人的医疗时代”,同时也引发了深刻的伦理思考。主要论证过程:1. 作者阿什利对基因组研究的热爱和使命感。2. 引用2021年末对2022年“黑天鹅”事件的预测,即基因技术延缓衰老、消除疾病,指出《基因密码》对此有翔实呈现。3. 介绍本庶佑的“先发制人的医疗时代”概念,即基于发病前诊断和预防性干预。4. 回顾基因和基因组研究的关键里程碑:基因是遗传基本单位、遗传密码破译、重组DNA技术(保罗·伯格)、全基因组关联分析(GWAS)、单核苷酸多态性(SNP)、CRISPR基因编辑技术(詹妮弗·杜德纳)。5. 强调CRISPR技术的革命性及其在遗传病治疗上的潜力。6. 引出核心议题:科技的善用与伦理困境。包括基因编辑婴儿事件、对“理想人类”的追求可能导致的社会趋同和对差异的不容忍(弗朗索瓦丝·贝利斯观点)、遗传决定论的风险(内森奈尔·康福特观点)。7. 以戴维·巴尔的摩的诗句作结,强调社会应如何使用改变人类遗传的能力。新奇的知识点:“先发制人的医疗时代”这一概念。CRISPR技术的通俗解释(基因剪刀)及其发现过程(细菌免疫系统)。对基因编辑伦理困境的深入探讨,引用了多位伦理学家和科学史学家的观点。前言 (作者:尤安·安格斯·阿什利)核心观点: 基因组是解开生命密码的关键,它不仅关乎人类本质和历史,更能预测未来健康与疾病,基因组学的飞速发展为医学带来了前所未有的机遇和挑战。主要论证过程:1. 作者自述对基因组的迷恋源于其巨大的科学潜力和帮助患者的成就感。2. 基因组的独特性和信息量:连接所有生命,包含个体历史和未来健康信息(身高、体重、疾病易感性等)。3. DNA的基本构成(A,T,G,C四种核苷酸)和规模(30亿碱基对,每个细胞DNA长2米,人体所有DNA可往返月球数千次)。4. 人类基因组计划的耗资与耗时,以及如今基因测序成本的急剧下降(法拉利价格从35万美元降至40美分的比喻)。5. 作者的个人成长经历:从小对医学和技术的兴趣,受《自私的基因》启发,最终选择心脏病学。6. 概述本书结构:早期基因组探索、基因诊断案例、心脏相关疾病的基因组研究、精准医疗的未来展望。7. 强调本书以患者及其家属为中心,致力于通过基因组研究改善他们的生活。新奇的知识点:同卵双胞胎基因组并非完全相同(胚胎形成后的变异、表观遗传修饰)。DNA双螺旋的右旋特性及其在科学插图中的常见错误。人类基因组中约10%来自远古病毒。作者用“法拉利降价”比喻基因测序成本的惊人下降。第一部分:早期基因组第1章:零号患者核心观点: 随着基因测序成本的急剧下降,对个体全基因组进行临床解读成为可能,预示着基因组医学时代的到来,但也带来了前所未有的医学和伦理挑战。主要论证过程:1. 以帕克·贝洛米(FOXG1综合征)的案例开篇,展示基因诊断的力量。2. 作者回忆2009年与同事斯蒂芬·奎克(物理学家、生物工程师)的会面,奎克向他展示了自己的基因组测序结果(Trait-o-matic软件)。3. 当时全基因组测序极为罕见且昂贵(克雷格·文特尔约1亿美元,詹姆斯·沃森约100万美元),而奎克用自研技术仅花费4万美元。4. 作者在奎克的基因组中发现一个与肥厚型心肌病相关的心肌肌球蛋白结合蛋白C(MYBPC3)基因变异体。5. 询问奎克的家族病史,发现其父亲有室性心动过速,堂兄的儿子19岁时猝死,这增加了基因变异体致病性的可能。6. 作者决定对奎克的整个基因组进行临床分析,奎克成为“零号患者”。7. 简述基因组基本知识:DNA构成(A,T,G,C)、染色体、基因(编码蛋白质,占2%)、非编码DNA(“垃圾DNA”的旧称)的重要性。8. 回顾DNA测序技术简史:弗雷德里克·桑格的测序法,以及“下一代测序”(NGS)技术如何实现大规模并行测序。9. 奎克使用的单分子测序技术(赫利克斯镜)及其成本和效率优势。新奇的知识点:Trait-o-matic:早期基因组表型关联工具。斯蒂芬·奎克成为早期接受全基因组临床解读的个体之一,其案例具有里程碑意义。假基因(Pseudogenes):基因组中与功能基因相似但失去功能的拷贝,可能对基因组分析造成干扰。单分子测序技术(赫利克斯镜)的工作原理。第2章:全明星团队核心观点: 对全基因组进行临床解读是一项复杂艰巨的任务,需要一个跨学科的“全明星团队”协同工作,并审慎处理伦理问题和知情同意。主要论证过程:排除了最初怀疑的心肌肌球蛋白结合蛋白C变异体的致病性。发现其冠心病(心肌梗死)风险高,与多种常见变异体和Lp(a)基因(环状结构重复段少导致Lp(a)水平高)相关。药物基因组学分析显示其对他汀类药物反应良好且副作用风险低。亨利·格里利:伦理、法律、社会影响专家(提出基因变异体健康影响的“交通信号灯”比喻)。阿图尔·布特:常见基因变异与疾病关联数据库,数据科学家团队。拉斯·奥尔特曼:药物基因组学知识库(PharmGKB)。作者团队:罕见变异体与罕见病。斯蒂芬·奎克团队:提供原始变异体列表。1. 组建团队应对基因组分析的三类挑战:大规模破坏性变异(单基因病)、常见变异体的累积风险(复杂疾病)、药物基因组学。2. 团队成员及其专长:3. 遗传咨询的重要性:凯莉·奥蒙德教授加入,处理知情同意、结果解读和“偶发性基因组”问题(“incidentalome”一词由扎克·科恩和拉斯·奥尔特曼提出)。4. 斯蒂芬·奎克的临床检查:听诊(勒内·拉埃内克发明听诊器)、心电图(奥古斯塔斯·沃勒与斗牛犬吉米的故事)、超声心动图、运动负荷试验,结果均显示心脏功能目前良好。5. 血液检查发现胆固醇问题:低密度脂蛋白(LDL,“坏”胆固醇)偏高,脂蛋白(a)[Lp(a)]水平极高。6. 团队历时6个月分析奎克的基因组:7. 基于基因组信息,团队建议奎克服用他汀类药物和阿司匹林,尽管ATP III等传统指南未能提供明确建议。8. 研究成果发表于《柳叶刀》(三篇论文),引起广泛关注,并在史密森尼博物馆展出。新奇的知识点:“偶发性基因组”(incidentalome)概念及其对全基因组测序的潜在影响。奥古斯塔斯·沃勒用斗牛犬“吉米”进行心电图实验的趣闻。脂蛋白(a)[Lp(a)]的遗传特性:基因大小(环状结构重复段数量)与Lp(a)水平成反比。首次将个体全基因组信息用于临床决策的详细过程。第3章:年轻人猝死之谜核心观点: 通过对死者进行全基因组测序(“分子尸检”),有可能揭示不明原因猝死的遗传因素,为家属提供答案,并帮助评估在世亲属的风险。主要论证过程:1. 讲述里奇·奎克(斯蒂芬·奎克的侄子)19岁猝死的故事,尸检未能明确死因。2. 其父瑞奇·奎克在悲痛中寻求答案,联系到堂弟斯蒂芬·奎克。3. 作者团队(加入弗雷德里克·杜威)决定对里奇进行“分子尸检”,从其心脏组织中提取DNA进行全基因组测序。4. 分析过程:建立可能导致心源性猝死的候选基因列表,比对里奇的基因组。5. 初步发现一个钾通道基因变异体,后证实是假基因(与真基因序列相似但无功能的DNA片段)干扰导致的误判,强调了基因组分析的复杂性。6. 最终锁定一个编码钙通道的基因变异体作为高度可疑的致病原因。7. 讨论死后基因检测的意义:不仅可能查明死因,更重要的是可以帮助评估和保护在世亲属(迈克尔·阿克曼和克里斯托弗·塞姆里安等人的研究)。8. 面临的挑战:死后基因检测的费用报销问题在美国医疗体系中存在障碍。9. 对比斯蒂芬·奎克(前瞻性风险预测)和里奇·奎克(回顾性死因探究)的案例,阐释基因组医学的不同应用方向。10. 引用达努尔杰·帕蒂尔关于模型迭代力量的名言,暗示基因组医学的持续发展。新奇的知识点:“分子尸检”(Molecular Autopsy)的概念及其应用。假基因对基因组分析,特别是短读长测序数据比对的干扰和挑战。即使是死后样本,全基因组测序仍可提供有价值的诊断信息。第4章:基因组测序,启航核心观点: 基因组测序技术的商业化,特别是索莱科萨(Solexa)公司的崛起及其被因美纳(Illumina)收购,极大地推动了测序成本的降低和技术的普及,为基因组学革命奠定了基础。主要论证过程:1. 索莱科萨公司的起源:1997年,剑桥大学的尚卡尔·巴拉苏拉曼尼安和戴维·克兰曼在潘顿酒吧讨论荧光标记核苷酸和单分子DNA测序,创立索莱科萨。2. 关键人物克莱夫·布朗的加入及其对早期技术的改进。3. 约翰·韦斯特的背景:麻省理工工程师,曾在伯乐公司(BioRad)和应用生物系统公司工作,了解DNA测序。4. 韦斯特被索莱科萨吸引的关键点:索莱科萨获得曼泰亚(Manteia)公司的DNA“簇”技术授权,能显著增强荧光信号,克服单分子测序信号弱的难题。韦斯特出任CEO。5. 索莱科萨的里程碑:成功对PhiX174噬菌体基因组进行测序。6. 商业策略:与濒临困境的上市公司林克斯治疗公司(Lynx Therapeutics)进行“反向合并”,在纳斯达克上市,并决定采用索莱科萨的DNA簇测序技术。7. 2006年推出首台商业测序仪(基因组分析仪GA),引起因美纳公司CEO杰伊·弗拉特利的注意。8. 索莱科萨为解决全基因组测序成本问题,计划开发靶向外显子组(基因编码区)测序技术,需大量寡核苷酸“诱饵”,因此联系因美纳。9. 2006年,因美纳以6.5亿美元收购索莱科萨,奠定了其在测序市场的主导地位。10. 因美纳后续推出GAIIx、HiSeq等高通量测序仪,进一步降低成本,并为10名个体(包括杰伊·弗拉特利和约翰·韦斯特及其家人)提供了个人基因组测序服务。新奇的知识点:索莱科萨/因美纳技术的诞生和发展历程,充满戏剧性的商业故事。DNA“簇”(cluster)技术对提高测序信号强度的关键作用。“反向合并”的商业操作。靶向外显子组测序概念的早期萌芽。第5章:首个家庭测序核心观点: 对核心家庭(父母和子女)进行全基因组测序,能更有效地揭示遗传规律、识别致病变异和评估家族成员的疾病风险,是基因组医学迈向临床应用的重要一步。主要论证过程:1. 约翰·韦斯特(前索莱科萨CEO,家人刚接受因美纳测序)联系作者,因其患有不明原因的复发性肺栓塞,担心遗传给孩子。2. 韦斯特17岁的女儿安妮已开始用Excel分析家庭成员的基因组变异识别文件(VCF),作为高中科学项目。3. 作者认识到家庭基因组分析的潜力:帮助确定遗传模式(显性、隐性、新生)、缩小致病变异搜索范围。4. 重组并扩大了分析斯蒂芬·奎克基因组的团队,新加入迈克·斯奈德(斯坦福遗传学教授,热衷多组学研究)和卡洛斯·巴斯塔曼特(斯坦福种群遗传学家)。5. 遇到的第一个难题:约翰·韦斯特临床诊断明确携带凝血因子V莱顿(Factor V Leiden)突变,但在其全基因组测序数据中未能找到该突变。新奇的知识点:高中生(安妮·韦斯特)参与分析家庭全基因组数据的超前案例。利用家庭成员基因组数据进行连锁分析和疾病基因定位的优势。凝血因子V莱顿突变在约翰·韦斯特基因组数据中“消失”的谜团,为下一章内容埋下伏笔。第6章:了解自己的基因核心观点: 人类参考基因组并非“完美”或“无疾病”的模板,其本身可能包含致病变异;创建基于多种群主要等位基因的参考序列能显著提高变异识别的准确性。同时,家庭基因组分析能揭示复杂的遗传模式和个体风险。主要论证过程:定相(Phasing):利用隐马尔可夫模型确定哪些等位基因共同遗传自同一亲本染色体。新生突变(De novo mutations):平均每个个体有40-80个新生突变。韦斯特家的孩子未发现重要新生突变。1. 人类基因组计划参考序列的构建历史:主要源自纽约州布法罗市罗斯威尔帕克癌症研究所的彼得·德容收集的匿名志愿者DNA(主要是RPCI-11,一位非裔美国男性,和RPCI-13,彼得·德容本人)。2. 解开约翰·韦斯特凝血因子V莱顿突变之谜:发现人类参考基因组在该位点本身就携带了莱顿突变。因此,韦斯特的正常等位基因被错误地识别为“变异”,而其致病的莱顿等位基因与参考序列一致,故未被检出。3. 这一发现揭示了严重风险:若患者对某个致病参考等位基因纯合,则可能漏诊。4. 弗雷德里克·杜威的解决方案:利用千人基因组计划数据,创建基于不同人群主要等位基因的新参考序列,提高变异检测的准确性。5. 家庭基因组分析方法:6. 阿图尔·布特团队为韦斯特家庭计算多基因风险评分,显示个体间风险差异,提示家族史并非完全可靠。7. 变异解读的人工工作与数据库建设:ClinGen联盟致力于标准化变异致病性分类。8. 向韦斯特家庭返还结果:约翰的凝血风险(遗传自他的莱顿因子V突变传给了女儿安妮),妻子朱迪也携带多种凝血相关变异,其中两种也传给了安妮,使安妮成为四种血栓风险变异的携带者。药物基因组学分析(华法林代谢正常,氯吡格雷超快代谢者)。新奇的知识点:人类参考基因组的真实来源及其包含致病变异的惊人事实。创建“主要等位基因参考序列”以提高变异检出准确性的方法。隐马尔可夫模型在基因组定相中的应用。多基因风险评分在家庭成员间的变异性。ClinGen联盟在变异分类和解读中的作用。第7章:从实验室到诊室核心观点: 将基因组测序和解读从学术研究推向临床应用,需要商业化运作、跨学科合作、伦理考量以及解决实际操作中的技术和成本挑战。主要论证过程:测序覆盖度问题:全基因组测序也可能漏掉医学重要基因的某些区域。成本效益:对“健康”个体测序后的额外医疗费用平均约700美元,并非天价。重要发现:一位参与者检出BRCA1突变(乳腺癌/卵巢癌高风险),凸显了基因组筛查的潜在价值和伦理沟通的重要性。1. 继奎克和韦斯特家族基因组分析后,临床基因组测序需求增加。作者与同事(奥尔特曼、斯奈德、布特)商议成立公司。2. 作者拜访硅谷传奇风险投资家迈克尔·莫里茨(红杉资本),阐述临床基因组解读公司的设想。3. 斯坦福大学鼓励技术商业化的文化背景(技术许可办公室,Bayh-Dole法案)。4. 邀请约翰·韦斯特(前索莱科萨CEO,也是基因组分析的亲历者)出任新公司CEO。5. 公司命名为“普森诺里斯”(Personalis),获得融资,创始团队庆祝。6. 迈克·斯奈德的“整合个人组学图谱”(iPOP)项目:对自己进行多组学(基因组、转录组、蛋白质组、代谢组、微生物组)长期监测,发现其TERT基因变异体(与再生障碍性贫血风险相关)和糖尿病发病。7. 斯坦福大学启动临床基因组测序试点项目(斯奈德和托马斯·奎特莫斯合作),面临批量解读基因组的挑战。8. 招聘遗传咨询师梅甘·格罗夫加入团队。9. 临床应用中的发现:10. 研究成果发表于《美国医学会杂志》(JAMA),提出基因组医学已从“诞生”走向“顽劣少年”阶段,需要规范发展。新奇的知识点:普森诺里斯(Personalis)公司的创立故事。迈克·斯奈德的iPOP项目,堪称极致的“量化自我”实践。全基因组测序覆盖度不均可能导致漏检医学重要变异。对健康个体进行基因组测序的后续医疗费用并非如预想般高昂。基因组医学发展阶段的“顽劣少年”比喻。第二部分:基因诊断第8章:未确诊疾病网络核心观点: 医学诊断,特别是针对疑难杂症,如同侦探工作,需要敏锐的观察力、系统的推理和多学科协作;美国国立卫生研究院(NIH)未确诊疾病网络(UDN)正是这种理念的体现。主要论证过程:1. 作者强调观察在医学诊断中的重要性,引用生活和临床中的例子(指甲、面色、步态等)。2. 将医学诊断比作夏洛克·福尔摩斯探案:柯南·道尔的医学背景,其导师约瑟夫·贝尔的观察力是福尔摩斯的灵感来源。福尔摩斯故事中充满了医学元素和观察推理。3. 现代流行文化中的体现:电视剧《豪斯医生》的创作灵感也来源于福尔摩斯。4. 介绍NIH的比尔·加尔医生,他长期致力于研究罕见代谢疾病,是现实版的“豪斯医生”。5. 加尔医生在2008年推动成立了NIH未确诊疾病计划(UDP),旨在解决最具挑战性的医学谜团。6. UDP最初面临资金和人手不足的困境,后得到NIH院长埃利亚斯·泽古尼和弗朗西斯·柯林斯的支持,逐渐发展。7. 2013年,UDP扩展为多中心的未确诊疾病网络(UDN),斯坦福大学成为首批临床站点之一,作者参与共同主持该网络。8. 将未确诊疾病患者的求医历程比作“医学奥德赛”,强调其漫长、曲折和充满不确定性。新奇的知识点:医学诊断与福尔摩斯探案手法的深刻类比。NIH未确诊疾病计划(UDP)及其发展为未确诊疾病网络(UDN)的历程和关键人物(比尔·加尔、埃利亚斯·泽古尼)。“医学奥德赛”这一术语形象地描述了疑难病患者的求医经历。第9章:寻找致病基因核心观点: 面对罕见遗传病,患者家庭的执着不懈、新兴的基因组测序技术(尤其是外显子组测序)以及互联网的连接力量,共同推动了新致病基因的发现。主要论证过程:1. 马特·迈特和克里斯蒂娜夫妇的儿子伯特兰患有不明原因的发育迟缓、无泪症和癫痫样发作。2. 经过漫长的“诊断奥德赛”,他们参与了杜克大学的外显子组测序研究项目。3. 2012年,杜克团队(范达纳·沙希等)将NGLY1基因的双等位基因失活突变确定为伯特兰的病因,伯特兰成为全球首例确诊NGLY1缺乏症的患者。4. 为寻找其他NGLY1患者以确证诊断,计算机科学家马特·迈特撰写了题为《追捕杀子凶手》的博客,并配上电影《飓风营救》中利亚姆·尼森的剧照,利用关键词优化和病毒式传播策略。5. 马特·威尔西和克里斯汀·威尔西夫妇的女儿格雷丝出现严重的肝脏问题和发育迟缓。6. 威尔西夫妇也经历了“诊断奥德赛”,分别在贝勒医学院和斯坦福大学(迈克·斯奈德团队)进行了外显子组测序。7. 贝勒医学院的博士后马修·班布里奇在研究格雷丝的基因数据时,通过谷歌搜索格雷丝的罕见症状(特别是无泪或少泪),找到了马特·迈特的博客。8. 格雷丝的症状与伯特兰高度相似,最终确诊格雷丝也患有NGLY1缺乏症。同时,土耳其一位医生也通过博客联系到迈特,其患者也被确诊。9. 多个家庭和研究团队通过互联网连接起来,共同确认了NGLY1是新的致病基因。新奇的知识点:马特·迈特利用博客和社交媒体“病毒式传播”寻找其他罕见病患者的创新方法。NGLY1缺乏症的发现过程,展示了外显子组测序在疑难病诊断中的早期应用和威力。两个独立家庭(迈特家和威尔西家)通过不同路径,最终因同一个罕见新基因病而联系起来的巧合与必然。第10章:对症下药核心观点: 明确致病基因后,深入理解疾病的分子机制,可能启发新的治疗策略,包括药物重用( repurposing)、补充剂治疗,甚至为未来的基因治疗和更精准的干预铺平道路。主要论证过程:1. NGLY1基因编码的N-聚糖酶1参与蛋白质去糖基化和细胞“垃圾回收”。功能障碍导致细胞内异常蛋白积聚。2. 马特·迈特了解到NGLY1缺乏症患者体内N-乙酰葡糖胺(GlcNAc)水平低,尝试从亚马逊购买GlcNAc补充剂给儿子伯特兰服用。3. 伯特兰服用GlcNAc后,首次流泪,其他症状也有所改善。犹他大学周恩慈团队的果蝇模型实验也证实GlcNAc能改善NGLY1缺陷果蝇的生存率。4. 诊断明确后对家庭生育计划的影响:可通过植入前遗传学诊断(PGD)或产前诊断避免将疾病遗传给下一代。5. 威尔西夫妇成立格雷丝科学基金会,资助NGLY1研究,连接患者家庭和科学家。6. 科学家铃木匡等发现抑制ENGase酶可能弥补NGLY1缺乏的影响。犹他大学团队通过计算机模拟筛选,发现质子泵抑制剂(如兰索拉唑)能抑制ENGase,伯特兰服用后进一步改善。7. 赫德森·弗里兹团队揭示NGLY1通过调控水通道蛋白(aquaporins)影响泪液分泌,解开无泪之谜。8. 强调基因诊断不仅带来答案,更带来希望、社群连接和推动治疗研究的动力。新奇的知识点:马特·迈特通过自行研究和网络购物,为儿子尝试补充剂治疗并取得初步成效的非凡故事。利用果蝇模型研究人类罕见病并测试治疗策略。药物重用:通过计算筛选发现已上市药物(质子泵抑制剂)对NGLY1缺乏症的潜在治疗作用。NGLY1缺乏症导致无泪的分子机制(与水通道蛋白相关)。患者倡导组织(如格雷丝科学基金会)在推动罕见病研究中的重要作用。第11章:探索未知病因核心观点: 未确诊疾病网络(UDN)通过整合全基因组测序、多学科专家会诊、功能实验验证(如iPSC、动物模型)以及传统的临床推理,成功诊断了大量疑难病例,发现了新综合征,并改善了患者管理。主要论证过程:全基因组测序发现ATP5F1D基因(线粒体复合物V亚基)双等位基因突变。通过国际合作(英国研究者罗伯特·泰勒在会议海报上发现相同基因),功能实验(iPSC来源的心肌细胞能量生成受损、线粒体形态异常)和果蝇模型验证,确认为一种新的线粒体病(MC5DN5)。基因组测序证实其同时携带FBN1(马方)和TRPS1基因的致病突变,是“五十亿分之一”的独特病例。诊断明确有助于针对性监测和管理。通过临床推理和文献查阅(福尔摩斯式方法),诊断为施尼茨勒氏综合征(一种罕见的自身炎症性疾病),而非基因组测序。治疗:白细胞介素-6(IL-6)阻滞剂有效。全基因组测序发现MECR基因双等位基因突变(MEPAN综合征),其中一个突变位于外显子组测序未覆盖的调控区。通过文献比对和临床表型匹配(语言运动迟缓但认知保留,基底节改变)指向诊断。治疗:补充α-硫辛酸和MCT油,病情稳定并改善。强调“撮合者交换”(Matchmaker Exchange)等平台对连接罕见病病例的重要性。UDN的优势:不受保险限制,可进行家庭测序,长期看更经济。1. UDN初期成果:对首批评估的382名患者诊断率达35%,发现31种新综合征,多数确诊病例的治疗得到改善。2. 案例一:米勒兄弟(卡森和蔡斯) - 运动神经发育迟缓,认知正常。3. 案例二:罗伯特·莱瑟斯 - 老年男性,长期不明原因发热、皮疹、肌痛。4. 案例三:阿莉莎·劳森 - 女孩,表现出马方综合征和毛发-鼻-指(趾)综合征(TRPS1)的混合特征。5. 案例四:安娜希 - 女孩,严重乳酸酸中毒,线粒体复合物功能障碍。6. 强调即使初步未诊断,科学进展和数据重分析也可能带来突破。新奇的知识点:MEPAN综合征的诊断过程,展示了全基因组测序相较于外显子组测序在检测调控区变异方面的优势。施尼茨勒氏综合征的诊断,体现了非基因组方法在特定病例中的价值。阿莉莎·劳森同时患有两种罕见遗传病的极端案例。利用iPSC诱导分化的心肌细胞和果蝇模型进行新疾病基因功能验证的完整流程。“撮合者交换”(Matchmaker Exchange)平台。第三部分:心脏那些事第12章:死亡线上跳舞核心观点: 快速全基因组测序技术的发展,使得在新生儿重症监护等危急情况下,能够迅速获得基因诊断信息,从而指导精准治疗,挽救生命。主要论证过程:迈斯纳(1856)描述聋哑女孩猝死。耶韦尔和朗格-尼尔森(20世纪50年代)描述伴耳聋的常染色体隐性LQTS(耶韦尔和朗格-尼尔森综合征,由KCNQ1基因突变导致,影响心脏和内耳钾通道)。罗马诺和沃德(20世纪60年代)描述不伴耳聋的常染色体显性LQTS(罗马诺-沃德综合征)。1. 新生儿杰兹琳因胎儿期心率过缓,出生后确诊为严重长QT间期综合征(LQTS),伴有致命性心律失常(尖端扭转型室性心动过速),多次心脏停搏。2. 回顾LQTS的发现史:3. 杰兹琳植入了当时世界上年龄最小的植入式心律转复除颤器(ICD)。4. 传统基因检测耗时太长(12周),作者团队联系因美纳公司,采用史蒂芬·金斯莫尔为新生儿重症监护室开发的50小时快速全基因组测序方案。5. 詹姆斯·普里斯特(作者实验室成员)协调样本递送和数据分析流程优化。6. 测序结果在几天内获得:杰兹琳携带KCNH2(hERG,钾通道)基因的一个已知致病变异(遗传自无症状父亲),以及RNF207基因(稳定KCNH2蛋白)的一个破坏性变异(遗传自无症状母亲)。7. 诊断为双基因遗传导致的严重LQTS,指导临床停用不必要的钠通道阻滞剂,重点加强针对钾通道异常的治疗。8. 加州大学戴维斯分校尼帕万·查维莫瓦特的细胞电生理实验证实了KCNH2和RNF207两个变异的协同致病效应,解释了杰兹琳病情严重而其父母无症状的原因。9. 杰兹琳一年后恢复良好。新奇的知识点:长QT间期综合征(LQTS)的详细发现史和不同亚型(伴/不伴耳聋,显/隐性遗传)。KCNQ1基因同时影响心脏和内耳功能的分子机制。新生儿快速全基因组测序在危重症诊断和治疗指导中的开创性应用。双基因(digenic)遗传导致更严重表型的案例。利用单细胞电生理实验验证基因变异功能的精细方法。第13章:我有两个基因组核心观点: 体细胞嵌合现象(个体拥有不同基因组的细胞群)可能导致严重的遗传性疾病,即便致病突变只存在于部分细胞中;检测嵌合现象需要特殊的细胞和分子技术,并可能需要创新的计算模型来理解其临床意义。主要论证过程:1. 继杰兹琳之后,另一名新生儿阿斯特里亚也因严重LQTS和心脏停搏入院。2. 快速全基因组测序提示SCN5A(钠通道)基因存在一个高度可疑的致病变异,但仅被一个生物信息学流程检出,且变异等位基因频率异常低(血液样本中约20%的读数支持变异)。传统桑格测序未能证实。3. 普森诺里斯公司对阿斯特里亚及其父母进行深度靶向测序,仍显示低比例变异(约8%),且父母均不携带该变异。4. 提出体细胞嵌合(somatic mosaicism)假说:阿斯特里亚体内部分细胞携带SCN5A新生突变。5. 介绍体细胞嵌合与嵌合体(chimerism)的区别及案例:普罗特斯综合征(象人)、节段性神经纤维瘤病、大脑半球巨大会、克隆性造血与心脏病风险、母亲携带胎儿细胞、异卵双胞胎血液嵌合、肾移植测试发现母亲有两个基因组的奇特案例。6. 为证实阿斯特里亚的嵌合现象,采用斯蒂芬·奎克发明的微流控单细胞DNA分析技术(富鲁达公司),发现其血液中确实约8%的细胞携带SCN5A突变。7. 阿斯特里亚病情恶化,心力衰竭,植入柏林心室辅助装置(VAD)作为心脏移植的过渡。8. 从其心脏活检样本中提取RNA进行测序,证实心脏组织中也存在SCN5A突变嵌合(5-12%),且分布不均。9. 为验证低比例嵌合是否足以致病,吉利德科学公司进行细胞实验,证实该SCN5A突变导致钠电流显著异常增强。10. 约翰斯·霍普金斯大学的纳塔利娅·特拉雅诺娃团队建立婴儿心脏计算模型,模拟不同比例和分布模式的嵌合细胞。最初模型(仅心肌细胞)未能复现LQTS。加入浦肯野纤维系统(心脏传导系统)后,模型成功模拟出LQTS、心脏传导阻滞和QRS增宽,与阿斯特里亚的临床表现一致。11. 阿斯特里亚成功接受心脏移植,健康成长。新奇的知识点:体细胞嵌合现象的详细解释和多种临床案例。首次将单细胞基因组分析技术(微流控)用于临床诊断嵌合现象。首次为婴儿心脏嵌合现象建立定制化的多尺度计算模型,并用于解释临床表型。心脏移植后患者可能同时成为嵌合体(原有嵌合)和嵌合体(移植器官)的复杂情况。第14章:“除之不尽”的肿瘤核心观点: 长读长基因组测序技术能够有效识别传统短读长测序难以检测到的大片段结构变异(如基因缺失),从而为复杂遗传综合征(如卡尼综合征)提供明确的分子诊断。主要论证过程:7岁时首次因右心房黏液瘤接受开胸手术。10岁时因睾丸肿瘤再次手术。临床高度怀疑卡尼综合征(以皮肤色素沉着、多发内分泌肿瘤和心脏黏液瘤为特征的遗传病)。卡尼综合征的发现史:爱尔兰病理学家J.艾丹·卡尼通过数十年观察,将看似不相关的症状联系起来,定义了该综合征。后由康斯坦丁·斯特拉塔基斯通过连锁分析确定PRKAR1A基因为主要致病基因。里基的PRKAR1A基因桑格测序结果为阴性。13岁时发现垂体瘤。16岁和18岁时因心脏肿瘤复发,再次接受开胸手术。后又因垂体瘤压迫和激素过量分泌,接受经鼻蝶垂体瘤切除术。1. 21岁的里基·拉蒙因心脏内多发良性肿瘤(黏液瘤)就诊。2. 里基的病史:3. 作者团队接手后,发现里基心脏肿瘤再次复发且生长迅速,面临第四次手术或心脏移植的抉择。里基最初犹豫,后因表姐意外去世并提出定向捐献心脏(虽未成功)而决定接受移植,但随后又撤出等待名单。4. 团队对里基进行全基因组测序(短读长),基因组数据管理员塔姆·斯内登注意到PRKAR1A基因起始区域覆盖度下降,怀疑存在缺失。5. 为验证此假设,采用太平洋生物科学公司(PacBio)的长读长测序技术对里基进行全基因组测序(首次临床应用)。6. 长读长测序明确检测到里基PRKAR1A基因一个拷贝存在超过2000个碱基的大片段缺失,解释了桑格测序阴性(因其检测的是剩余的正常拷贝)和卡尼综合征的临床表现。7. 里基病情危急,在移植会议上,心胸外科主任约瑟夫·吴决定为其进行第四次高风险开胸手术,切除多个肿瘤并重建右心房和上腔静脉。术中发现三尖瓣严重反流,再次修复。里基术后恢复迅速。新奇的知识点:卡尼综合征的详细发现史,特别是J.艾丹·卡尼医生如侦探般的临床观察和不懈追踪。PRKAR1A基因作为卡尼综合征主要致病基因的确定过程。首次将长读长全基因组测序技术成功应用于临床,诊断出由大片段基因缺失导致的孟德尔遗传病。里基·拉蒙多次经历重大心脏手术和复杂决策过程的感人故事。第15章:失忆式假死核心观点: 肥厚型心肌病(HCM)是一种常见的遗传性心脏病,其认识历程从病理观察到临床诊断,再到基因层面的解析,极大地改变了患者及其家庭的命运和管理策略。主要论证过程:克里斯蒂娜·塞德曼和乔纳森·塞德曼夫妇(与比尔·麦克纳合作)通过对一个魁北克大家族(科蒂库克镇,最早由J.A.P.帕尔研究)进行连锁分析,于1990年首次确定MYH7(心肌肌球蛋白重链)基因为HCM致病基因。随后发现更多致病基因,如MYBPC3(心肌肌球蛋白结合蛋白C)等。17世纪博内首次描述“公牛般的心脏”。18世纪莫尔加尼描述斑片状纤维化和室间隔肥厚,兰西西记录家族遗传。20世纪50年代,唐纳德·蒂尔通过大量尸检详细描述了HCM的病理特征(心肌细胞排列紊乱),并将其与临床表现联系起来。保罗·伍德(著名心脏病学家)总结了HCM的临床体征。20世纪60年代,安德鲁·格伦·莫罗(尤金·布朗沃尔德合作者)开创了室间隔心肌切除术(莫罗氏手术)治疗梗阻性HCM,莫罗本人后也死于HCM。超声心动图技术的出现彻底改变了HCM的活体诊断,可观察到特征性的“收缩期前向活动”(SAM)。巴里·马龙和威廉·麦克纳在HCM研究领域贡献卓著。1. 13岁的莉拉妮·格雷厄姆在跑步机上运动时突发心脏骤停,后确诊为肥厚型心肌病(HCM)。2. HCM的历史回顾:3. 莉拉妮心脏骤停后的短期失忆(顺行性遗忘)现象及其神经生理学基础(海马区对缺氧敏感)。4. 莉拉妮植入ICD后,仍经历了两次心脏骤停(体育课跑步、爬土坡)。5. 作者团队在她17岁时接手,建议进行HCM相关基因检测。6. HCM的遗传学基础:7. 莉拉妮的基因检测结果:MYBPC3基因存在两个不同的致病性变异,分别遗传自无症状的父母。8. 其妹妹卡伊接受基因检测,未遗传到任何致病变异,解除了患病风险。9. 莉拉妮前往纽约大学学习音乐剧,在街头再次发生心脏骤停。新奇的知识点:肥厚型心肌病(HCM)的详细发现和认识发展史,包括多位关键医学人物的贡献。心脏骤停后顺行性遗忘的机制。首个HCM致病基因MYH7的发现过程,利用了连锁分析和大家系研究的经典遗传学方法。基因检测在HCM诊断、家族筛查和风险评估中的重要作用及对家庭的情感影响。第16章:移植“坏”心脏核心观点: 心脏移植是终末期心力衰竭的有效治疗手段,但术后可能出现新的复杂问题,甚至需要对移植心脏本身进行基因组学探究;同时,对HCM等疾病分子机制的深入研究(如肌球蛋白功能异常)为开发新的治疗靶点提供了基础。主要论证过程:斯坦福大学的詹姆斯·斯普迪赫(分子马达研究专家)和莱斯莉·莱恩万德(肌球蛋白制备)合作研究HCM相关肌球蛋白突变。斯普迪赫通过体外实验(肌动蛋白丝滑动实验、激光陷阱)发现HCM突变不一定都增强肌球蛋白的单分子力量。斯普迪赫的“方山”(mesa)理论:肌球蛋白头部存在一个平台状结构(方山),是与其他蛋白(如MYBPC3)相互作用的关键区域。HCM突变多发生于此,可能通过影响这种相互作用,导致更多肌球蛋白分子处于“激活”状态(“更多的桨在划水”),从而引起心肌过度收缩。作者实验室的朱利安·洪布格尔通过对大量人群基因数据和肌球蛋白三维结构进行建模分析,证实HCM致病突变确实在“方山”和“转换器”等关键功能域富集,支持了斯普迪赫的理论。1. 莉拉妮从纽约大学转至南加州大学后,HCM病情进展,出现心力衰竭症状,经多方评估后列入心脏移植等待名单。2. 肥厚型心肌病协会(莉萨·索尔伯格创立并领导)为患者提供支持。莉萨本人也是HCM患者和心脏移植受者。3. 莉拉妮成功接受心脏移植手术,但术后新心脏出现严重电传导问题,反复停搏,需要ECMO支持。4. 其父克里斯·格雷厄姆(MYBPC3变异携带者)在巨大压力下出现室性心动过速,植入ICD。5. 为探究莉拉妮新心脏问题,团队决定对其移植心脏的基因组进行测序(通过心肌活检样本)。6. 测序结果初步显示新心脏也携带其原有的MYBPC3变异,后确认为样本中混有莉拉妮自身细胞所致。通过“减去”莉拉妮自身基因组信息,发现移植心脏GJA5(间隙连接蛋白40)基因存在一个与心脏传导阻滞相关的变异。莉拉妮对此“并不意外”。7. 探讨HCM的分子机制:8. 莉拉妮凭借对艺术的热爱和患者倡导工作,继续书写人生。新奇的知识点:对移植心脏进行基因组测序以探究术后并发症的罕见案例。GJA5(间隙连接蛋白40)基因变异与心脏传导问题的关联。詹姆斯·斯普迪赫关于HCM分子机制的“方山”理论和“桨与船”比喻,为理解HCM的超收缩状态提供了新视角。结合群体遗传学数据和蛋白质三维结构分析来验证疾病分子机制假说的方法。第四部分:迈向精准医疗第17章:超人基因核心观点: 研究那些拥有罕见“超人”基因(如赋予极强耐力、极低胆固醇或痛觉缺失)的个体,不仅能揭示人体生理极限的奥秘,更能为常见疾病(如贫血、心脏病、慢性疼痛)的治疗提供新的药物靶点和研发思路。主要论证过程:这提示SCN9A是开发新型非阿片类镇痛药的理想靶点。但由于SCN9A与其他钠通道亚型高度同源,开发高特异性抑制剂面临挑战。药企开始探索抗体药物或天然产物(如狼蛛毒液)作为解决方案。PCSK9基因的发现史:凯瑟琳·布瓦洛等发现PCSK9功能获得性突变导致家族性高胆固醇血症。海伦·霍布斯和乔纳森·科恩在达拉斯心脏研究中发现,非洲裔人群中存在PCSK9功能缺失性突变(莎拉妮为纯合缺失),导致LDL极低,并显著降低心脏病风险。这一发现直接催生了PCSK9抑制剂类降脂新药(如依洛尤单抗、阿莫罗布单抗等抗体药物)的研发。1. 埃罗·门蒂兰塔案例: 芬兰传奇越野滑雪奥运冠军,拥有异常高的红细胞比容(60-70%)。遗传学研究(阿尔贝特·德·拉卡佩勒)发现其家族携带促红细胞生成素(EPO)受体基因突变,导致骨髓对EPO高度敏感,红细胞生成旺盛,赋予其超强耐力。2. 作者团队的“精英”(ELITE)项目:研究世界顶级耐力运动员的基因组,寻找“超人基因”。例如,发现有运动员携带与安第斯人适应高原相关的基因变异,或与细胞能量代谢(NAD通路)相关的有利变异。3. 莎拉妮·特蕾西案例: 非裔美国女性,LDL(“坏”胆固醇)水平极低(14 mg/dL)。4. 孟德尔随机化法: 利用基因变异的随机分配特性,判断观察到的关联是因果关系还是仅仅相关。例如,证明了低LDL(而非高HDL)与心脏病风险降低之间存在因果关系,避免了药企在无效靶点上的巨大浪费。5. 先天性痛觉缺失症案例: 巴基斯坦某家族成员因SCN9A(一种钠通道)基因双等位基因失活突变而感觉不到疼痛。6. 结论:研究“超人”基因有助于验证药物靶点,加速新药研发。新奇的知识点:埃罗·门蒂兰塔的EPO受体基因突变赋予其“天然血 doping”的生理优势。PCSK9基因功能缺失赋予部分人群天然的“降脂保护”。孟德尔随机化法作为一种利用遗传学信息推断因果关系的强大工具。SCN9A基因与痛觉感知,及其作为非阿片类镇痛药靶点的潜力与挑战。从天然毒液(如狼蛛毒液)中寻找药物先导化合物的策略。第18章:精准医疗核心观点: “精准医疗”旨在整合个体的基因组信息、生活方式、环境因素和健康数据,以实现更个性化的疾病预防、诊断和治疗。大规模人群队列研究(如英国生物样本库、“我们所有人”项目)是推动精准医疗发展的基石。主要论证过程:案例:西奥多·卡特,一名运动员,因MYH7一个变异被误诊为HCM并限制运动。gnomAD数据显示该变异在人群中并不少见,推翻了原诊断。基因解码公司(deCODE,冰岛,卡里·斯特凡森领导):利用冰岛独特人群进行大规模遗传学研究,成果卓著。开发了“冰岛人之书”数据库和有趣的“防近亲约会”App。英国生物样本库(UK Biobank):50万参与者,数据向全球研究者开放,已产出上千篇研究论文,极大推动了心脏病、癌症、神经系统疾病等多领域研究。作者团队利用其数据研究主动脉瓣二瓣化畸形的遗传基础。英国计划进一步扩大基因组测序规模。直接面向消费者的基因检测公司(如“染色体和我”、族谱网)积累了海量用户数据,并开始涉足药物研发。其他大型队列:中国慢性病前瞻性研究、百万老兵计划(显示了多种族队列在发现新关联上的优势)。吉尔·霍尔德伦(总统科技顾问约翰·霍尔德伦之女,BRCA1突变携带者)与奥巴马总统的会面,分享其经历和对基因组医学的见解,对总统产生深刻影响。奥巴马指示白宫科技政策办公室制定相关提案。2015年国情咨文,奥巴马正式宣布启动“精准医疗倡议”,强调为患者提供正确时间、正确治疗,并让患者获取自身数据。弗朗西斯·柯林斯(NIH院长)2003年即设想建立美国大型人群队列,但因成本和认知受阻。贝拉克·奥巴马任参议员时(受多拉·休斯、詹妮弗·莱布等人影响)即关注个性化医疗,2007年提出《基因组和个性化医疗法案》(未通过)。1. 埃里克·迪什曼的经历: 19岁被误诊为罕见肾癌,预后极差。后肿瘤基因组测序揭示其更像胰腺肿瘤,调整化疗方案后病情缓解,最终成功接受肾移植。其经历促使其成为以患者为中心的精准医疗的倡导者。2. 精准医疗理念的早期萌芽:3. 美国精准医疗倡议(PMI)的诞生:4. PMI的关键组成:癌症研究、FDA合作(推动基因检测审批)、建立百万人国家队列研究(后命名为“我们所有人”项目)、保护个人隐私。5. “我们所有人”(All of Us)研究项目: 埃里克·迪什曼被任命为负责人,项目强调受试者作为合作伙伴,注重人群多样性,并承诺向受试者返回健康和遗传信息。6. 国际大型队列研究的进展与影响:7. 数据整合与共享的价值: gnomAD数据库(丹尼尔·麦克阿瑟领导)整合了全球数十万人的外显子组和基因组数据,为罕见变异的频率评估和致病性判断提供了关键资源。新奇的知识点:埃里克·迪什曼从癌症患者到精准医疗倡议领导者的传奇经历。美国精准医疗倡议(PMI)和“我们所有人”(All of Us)项目的背景、目标和运作模式。冰岛deCODE公司利用系谱数据开发的“防近亲约会”App。英国生物样本库的开放数据模式及其对全球科研的巨大贡献。gnomAD数据库在临床基因变异解读中的革命性作用,及其如何帮助纠正错误诊断。“精准医疗”一词的由来(美国国家科学研究委员会2011年报告)。第19章:基因治疗核心观点: 基因治疗在经历了早期挫折后,凭借病毒载体技术的改进、RNA干扰等新策略以及CRISPR等基因编辑工具的出现,正迎来黄金时代,为多种遗传性疾病(如血友病、脊髓性肌萎缩、遗传性失明)带来了前所未有的治愈希望。主要论证过程:米拉·马科维茨(巴滕病,CLN7基因突变)案例:波士顿儿童医院游维文团队通过全基因组测序发现其一个拷贝存在独特的“跳跃基因”插入。团队为米拉量身定制了反义寡核苷酸药物“米拉森”(milasen),靶向校正跳跃基因导致的剪接缺陷。一年内从诊断到治疗,米拉病情显著改善。CRISPR/Cas9系统的发现(莫伊察、西克斯尼斯、沙尔庞捷、杜德纳、张锋、丘奇等人的贡献),彻底改变了基因编辑领域。刘如谦团队开发的单碱基编辑器和先导编辑器(prime editing)进一步提高了编辑精度和范围,无需DNA双链断裂。应用前景广阔,但人类胚胎基因编辑的伦理问题引发巨大争议(如米塔利波夫的人类胚胎HCM基因编辑研究)。安德鲁·费尔和克雷格·梅洛发现短双链RNA可特异性关闭基因表达(诺贝尔奖成果)。挑战:RNA分子的体内递送。通过脂质纳米颗粒(LNP)包裹递送至肝脏取得突破。Onpattro(Patisiran):首个获批的RNAi药物,用于治疗遗传性转甲状腺素蛋白淀粉样变性,能逆转疾病进展。Inclisiran:靶向PCSK9的RNAi药物,用于降胆固醇,每年仅需注射两次。作者实验室在HCM等位基因特异性沉默方面的研究。SMN1基因突变导致。传统上无有效疗法。反义寡核苷酸疗法(Nusinersen/Spinraza):通过鞘内注射,调控SMN2基因的剪接,使其产生更多功能性SMN蛋白,显著改善运动功能和生存率。AAV基因替代疗法(Zolgensma/Onasemnogene abeparvovec):单次静脉注射,将正常SMN1基因递送至运动神经元,效果显著。莱伯先天性黑矇(RPE65基因突变):克里斯蒂安·瓜尔迪诺等患者接受Spark Therapeutics公司的基因治疗(Luxturna,AAV载体将正常RPE65基因注射至眼底),视力显著改善。眼睛因其“免疫赦免”特性成为基因治疗的理想靶点。血友病B(凝血因子IX缺乏):通过AAV载体递送高活性“帕多瓦”变体因子IX基因,实现持续的治疗水平。血友病A(凝血因子VIII缺乏):基因较大,递送困难。通过优化AAV载体和使用缩短版因子VIII基因,在临床试验中取得显著疗效,大幅减少出血事件。20世纪90年代初首次临床应用,但效果不佳,安全性问题突出(1999年杰西·格尔辛格死亡事件,SCID基因治疗导致白血病)。关键挑战:基因的有效、安全递送。病毒(尤其是腺相关病毒 AAV)因其天然的基因递送能力成为主要载体。1. 以作者同事、已故肿瘤学家霍尔布鲁克·科尔特(患有血友病A)的故事引出血友病的治疗困境和对基因治疗的期盼。2. 基因治疗的早期历史与挑战:3. 血友病的基因治疗突破:4. 遗传性失明的基因治疗:5. 脊髓性肌萎缩(SMA)的基因治疗:6. 基因沉默(RNA干扰,RNAi):7. CRISPR基因编辑:8. N-of-1个性化基因治疗:新奇的知识点:霍尔布鲁克·科尔特的感人故事。血友病、莱伯先天性黑矇、脊髓性肌萎缩等疾病基因治疗的最新进展和代表性药物(Luxturna, Spinraza, Zolgensma, Onpattro)。RNA干扰(RNAi)的原理及其在药物开发中的应用(如靶向TTR和PCSK9)。CRISPR/Cas9基因编辑技术的发现历程、关键人物及其革命性影响,以及单碱基编辑、先导编辑等更精确的技术。“米拉森”(milasen)作为首个针对个体独特基因突变定制的“N-of-1”基因治疗药物的里程碑意义。第20章:前方的路核心观点: 基因组医学的未来将以更快、更准、更便宜的测序技术为基础,结合人工智能、大数据分析和多组学方法,实现对常见病和罕见病的精准预测、预防和个体化治疗,并在公共卫生(如传染病监测与应对)领域发挥关键作用,但同时也面临技术、伦理和经济上的持续挑战。主要论证过程:克服基因治疗药物向更多靶器官(如肌肉、肺、神经系统、心脏)有效递送的挑战。利用CRISPR等基因编辑技术直接对抗病毒或修正致病基因。解决基因治疗药物的高昂成本和公平可及性问题,以及相关的伦理和社会考量。实时病原体基因组测序在传染病暴发(如COVID-19大流行)中的关键作用:快速鉴定病原体、追踪传播途径、加速疫苗(如mRNA疫苗、病毒载体疫苗)和诊断方法(如CRISPR检测)的开发。利用城市污水中的微生物基因组进行传染病早期预警。多基因风险评分(PRS)将更准确,并整合不同种族人群数据,用于心脏病、癌症等疾病的风险分层和筛查指导。与生活方式、环境因素、可穿戴设备监测数据结合,实现更个性化的预防策略。长读长测序(如PacBio)和纳米孔测序(如Oxford Nanopore,克莱夫·布朗领导)将提高结构变异检测能力、简化基因组组装、实现DNA/RNA化学修饰(表观遗传)的直接读取,并进一步降低成本、提高速度、增强便携性(如用于现场病原体检测、粮食安全)。1. 测序技术的持续进步:2. 常见病的精准医疗:3. 基因组学与公共卫生:4. 基因治疗的未来:5. 作者回顾过去十余年基因组医学的飞速发展,从少数个体的研究探索到日益普及的临床应用,展望了充满希望但也挑战重重的未来。新奇的知识点:纳米孔测序技术的特点(便携、实时、长读长、可检测化学修饰)及其在现场检测和公共卫生中的应用潜力。多基因风险评分(PRS)在常见病预防中的应用前景和种族多样性挑战。利用污水宏基因组学进行传染病早期预警的创新思路。COVID-19大流行如何加速了基因组学技术(测序、CRISPR诊断、基因疫苗)的应用和发展。基因治疗面临的递送难题和成本挑战。致谢核心观点: 作者向所有在其研究和写作过程中给予帮助和启发的人们表示感谢,特别是患者及其家属、科研合作者、导师、家人以及出版团队。主要论证过程: 逐一致谢。
基因疗法
2024-06-24
近日,Exicure收到纳斯达克股票市场上市资格部的退市决定通知,由于其提交截至2023年12月31日的2023年财年的10-K表格,不符合继续上市的要求。
此外,纳斯达克股票市场上市资格部还指出,Exicure未能举行2023年年度股东大会、以及按时提交2024年第一季度的10-Q表格同样违反了纳斯达克交易市场继续上市规定。
曾经,为了不触及“1美元退市红线”,Exicure采取了反向分割股权手段。如今,公司却数条上市合规而遭到退市。
注:10-K是美国证券交易委员会(SEC)要求上市公司每年提交的有关其财务业绩的综合报告(经审计)。
10-Q为每季度必须向SEC提交的财务业绩综合报告(通常未经审计),每年只需提交三次。
反向股票分割是上市公司为了减少其在市场上的流通股数量而采取的一种措施。在反向股票分割过程中,公司注销其当前的流通股,并按照反向分割前股东持有的股份数量向股东分配新股。
投资大佬云集
Exicure是核酸药物开发领域的明星公司,于2015年成立,2019年在纳斯达克上市,致力于开发治疗肿瘤、炎症以及遗传病的核酸药物。
Exicure是球形核酸(SNA™)药物的先驱,其核心技术-球形核酸(SNA)纳米技术以人造纳米球作为支架,将单链或双链核酸装载在纳米球表面。这种形态的核酸可以在不触发免疫系统的情况下顺利进入细胞,同时球形结构可以将细胞对核酸的摄取提高到线性核酸的1000倍。
凭借这一技术,Exicure吸引了一些诸多世界知名投资大佬的青睐,早期投资者包括Bill Gates(微软创始人)、David Walt (Illumina创始人)、George Rathmann (Amgen创始人)、Eric Schmidt (Google创始人),另外,国内的绿叶制药集团也是投资方。
2019年11月,Exicure成功将一款治疗脱发管线授权Allergan(被艾伯维收购),交易总额高达7.25亿美元(2500万美元的首付款及未来的里程碑付款)。
在这些利好的推动下,Exicure的股价曾最高一度达到115.2美元。
数据造假丑闻
然而2021年的数据造假事件给予了公司最沉重的一击,并引发了市场对公司的信任危机。
2021年11月,Exicure的一项内部调查发现,其神经科学部门的负责人在至少三项关键性临床前试验的数据中造假,使得公司对于XCUR-FXN的有效性出现了误判。
虽然Exicure声称公司其他人对此并不知情,更没有任何人参与造假事件,但仍造成了市场对公司信任的崩塌。该消息公布后,Exicure的股价一泄千里,从30多美元一路跌至5美元。
随之,Exicure采取了重组措施,裁掉了50%的员工,砍掉两条管线XCUR-FXN和AST-008,SCN9A被提为公司研发的最高优先级,重点推进研发工作。
重组之后,Exicure依旧动荡不止。2022年2月,CEO、CTO以及三名董事会成员先后离开公司。而首推的管线SCN9A也令人失望,在非人灵长类模型中,SCN9A作为非阿片药物的替代品治疗神经性疼痛未能达到之前在体外临床研究中的效果。公司随即暂停了SCN9A的研发工作。
这时,Exicure的财务状况也十分堪忧。2022年半年报显示,公司营收503.6万美元,净亏损-1581万美元,公司账上现金仅剩2000万美元,仅供维持半年时间。
2022年9月,Exicure宣布更彻底的重组,裁员66%,停止所有研发工作,探索最大化股东价值的战略替代方案。同时,Exicure以540万美元的对价,将公司50.4%的控股权转让给一家名叫CBI USA的投资公司,也是凭着这笔资金,Exicure能够运转至今。
但是现如今Exicure的最主要目标,就是做好准备,等待一个愿意出手的买家。
参考出处:
https://www.biospace.com/article/releases/exicure-inc-received-nasdaq-notice-of-a-delisting-determination/
https://investors.exicuretx.com/news/news-details/2021/Exicure-to-Present-at-Upcoming-Scientific-Conferences-27feffa01/default.aspx
https://firstwordpharma.com/story/5686393
https://exicuretx.com
数据误报,还是学术作假?上市新星深陷寒潭
本周好文推荐
如需转载请联系佰傲谷并在醒目位置注明出处
·
·
·
·
·
·
·
·
并购高管变更
2022-11-28
·药智网
为了纪念令人毛骨悚然的生物技术市场,Fierce Biotech于近日推出了2022年踏入墓地的生物科技公司名单。该名单共有7位成员,分别是Diffusion制药、Exicure、Genocea、H3 Biomedicine、Kaleido、Orphazyme、Bone。其中Exicire和Diffusion制药两家公司尚未完全闭气,只是因为已经大幅缩减研发工作,半身子躺在棺材里,所以“上榜”。某种程度上来说,该名单不只是市场的茶余饭后的谈资。因为,这是一份在金融动荡和临床失败的危险重压下,崩溃的biotech名单。导致这些企业失败背后的某些“经历”,或许能够为整个行业带来“警示”作用。01Diffusion制药——缺乏“出售”自己的能力对于Biotech来说,融资能力也是实力的体现。缺乏融资能力,或者说没有“出售”自己能力的Biotech,危险系数无疑更高。Diffusion制药,便诠释了这一点。Diffusion制药是一家研发“组织缺氧”药物的biotech。公司的TSC疗法,能够在低氧的条件下改善组织供氧,目前已开展了肺炎、贫血和实体瘤等多种适应症的临床试验。Diffusion制药的首席执行官Robert Cobuzzi Jr博士表示,TSC也可以作为多形胶质母细胞瘤等低氧实体肿瘤的辅助治疗。但前提是,公司能找到一笔交易或其它合作伙伴来帮助它度过这场危机。的确如此,Diffusion制药已经捉襟见肘多时。截至2022年9月末,公司帐上现金只有2585万美金。在寒冬中,公司的市值跌至1000万美金,基本丧失了再融资能力。今年10月,Diffusion制药宣布,公司可能会进行合作、许可交易、剥离甚至完全出售。不过,各项计划也可能只是说说而已。目前,这些项目没有完成的时间表,管理层也没有承诺何时会有新消息。02Exicure——输给“内控”的公司一直以来,科学家能否经营公司是市场最大的疑惑。核心原因在于,对于一家公司来说,核心竞争力不仅包括研发,还包括管理、内控等。Exicure或许就是一家败给“内控”的公司。Exicure是一家专注于神经系统疾病创新疗法研究的biotech。这家诞生于美国芝加哥的biotech,含着金汤匙出生,从比尔·盖茨等人那里筹集了数千万美元。完成IPO后,市值一度逼近10亿美金,成为“准独角兽”。遗憾的是,Exicure经常因为各种错误登上头条。公司最大的问题,是“内控”。一项内部调查发现,Exicure的前神经科学小组组长,在2021年前9个月里误报试验数据。这直接导致Exicure放弃了核心管线的研发。从那以后,公司厄运连连,一直在削减管线和裁员,并调整研发方向。在遭遇挫折之后,Exicure将研究重点转移至临床前疼痛项目SCN9A。然而,研究人员在动物实验中发现,该候选项目不合格,公司随即暂停了所有的研发工作。在此期间,Exicure进行了内部重组,并进行裁员以削减成本并调整业务、精简管线。始终在动荡中蹒跚前行,Exicure如今的市值只剩下区区561万美金。今年9月底,CBI公司以540万美元的价格,购买公司340万股普通股,这才让Exicure能够运转至今。03Genocea——资金耗尽导致散伙在资本寒冬中,Biotech最大的挑战,是资金。对于大部分biotech来说,需要推进管线研发以获得市场认可,而没有资金意味着难以完成上述动作。这将导致,即便你自认为实力出众,但也无法让市场意识到你真的实力出众。Genocea便是在这一背景下,踏入墓地的biotech。Genocea自以为傲的,是其专有的发现平台ATLAS。根据公司表述,ATLAS平台可以使用患者自身的T细胞免疫反应机制来识别最佳抗原,从而实现更加个性化的癌症免疫疗法。基于该平台,公司开发了一款肿瘤疫苗,和特定新抗原的T细胞疗法。正是凭借技术平台+管线的故事,Genocea在二级市场市值一度超过14亿美金。遗憾的是,公司没能证明自己,导致难以获得再融资。从2013年至今,公司帐上现金始终在数千万美金徘徊,亚历山大。在经历了多年的财政压力后,Genocea于5月24日正式宣布退市,并遣散了剩余的非必要员工。Genocea一直试图坚持下去。2022年4月,Genocea宣称在研T细胞疗法产品GEN-011在I/IIa期临床试验中展现了一定疗效,5名患者中4名病情稳定,并计划下一阶段研究中继续增加剂量。然而,仅仅几周后,Genocea就宣布了一项战略评估,旨在出售公司的全部或部分资产并裁员65%。最终结局,如上文所说,散伙。04H3 Biomedicine——大药厂缩编的一个缩影近年来,大药厂频频调整,子公司裁撤无数。关门大吉的H3 Biomedicine,就是大药厂减员的一个缩影。H3 Biomedicine是日本药厂卫材的子公司,成立于2010年,位于美国马萨诸塞州剑桥,专注于发现和开发精准肿瘤治疗方法。自2010年成立以来,H3一直是重要卫材的创新发现平台和开发引擎,目前已经提交了4项IND,签署了2份授权协议,完成了H3B-6545概念的临床验证。”不过,近年来跨国药企在市场压力之下调整不断,卫材便是其中一员。今年7月,卫材决定永久关闭其肿瘤学部门H3 Biomedicine,裁员人数预计为88名。其将全球研发(R&D)组织从以前的业务集团结构改革为新的研发组织DHBL。H3 Biomedicine在研业务也将合并到DHBL组织中。对此,市场猜测可能是由于投入成本超出预期、肿瘤线高管变动、人员有所调整、研发进度存在问题,也可能是卫材认为阿尔兹海默症药物未来的市场潜力大,做了资源的优先倾斜。05Kaleido——坐拥全球顶流资源也难逃失利在生物科技行业,大佬坐镇是加分项,但也仅仅只是加分项而已,并不能代表实质性的利好。历史上,大佬失利的例子比比皆是。Kaleido也是这样一个例子。看上去,Kaleido是一家玄学公司,其宗旨是以“解锁微生物群,改变医药世界”。不过,Kaleido背景绝对够硬。其为顶级投资机构Flagship Pioneering孵化的明星,与Moderna属于同门师兄弟。成立之后,Kaleido与诸多大佬搭上关系,包括与全球著名的MD安德森癌症中心的医学博士Robert Jenq合作,与慢性阻塞性肺病基金会联手,和强生联姻探索微生物组代谢疗法在预防特应性疾病、免疫疾病。公司C轮融资达到1.01亿美金,且在IPO后市值超过10亿美金。不过,由于迟迟没有拿出成绩,投资者兴趣衰退,公司逐渐陷入危机。这种背景下,公司甚至铤而走险。Kaleido核心产品KB109是一款治疗多重耐药性细菌感染的靶向聚糖,在未经FDA许可的情况下,意图用于缓解和治疗新冠肺炎。对此,FDA显然不接受,下发警示函终止公司的荒诞行为。这,也阻断了Kaleido最后的生路。今年2月,面对日益减少的现金储备,KB109用于慢阻肺的II期临床研究被迫终止。到了4月份,在未能找到买家,且资金“败光”的情况下,Kaleido宣布“无法继续运营,最好的选择是有序地结束流程”,正式宣告踏入墓地。06Orphazyme——被FDA劝退的明星选手小型生物技术公司通常依赖一两种产品,这使它们成为高风险、高回报的投资类型。Orphazyme是一家“高风险”的案例。Orphazyme是一家位于丹麦的biotech,专攻神经退行性疾病药物研发。其核心管线arimoclomol旨在治疗一种罕见的进行性遗传疾病C型尼曼-匹克病。该疾病适用人数极少,全球患者总数估计约为3000人;但商业化前景可能不低,公司预计该疗法治疗价格约为30—60万美金。在这一背景下,Orphazyme二级市场巅峰市值逼近30亿美金。遗憾的是,arimoclomol的临床数据显示,该药物疗效并不明显。最终arimoclomol被FDA和EMA接连劝退。受这些负面消息影响,Orphazyme股价持续暴跌,市值只剩下3000万美金。更悲剧的,核心管线失利,导致Orphazyme陷入裁员风波,最终破产。Orphazyme的结局,是被KemPharm以不到1300万美元的价格收购。头铁的KemPharm表示,还将继续推进arimoclomol上市申请的计划。在此,祝KemPharm好运。07Bone——摊子铺太大对于Biotech来说,管线越多并不意味着越好。毕竟,初创型企业有没有能力推进诸多管线存疑,单单资金压力就不小。Bone Therapeutics或许就是摊子铺太大。Bone Therapeutics是一家骨细胞治疗公司,专门解决骨科和骨病领域未满足的医疗需求。根据介绍,公司拥有广泛的创新同种异体细胞治疗解决方案组合,涵盖一系列适应症。其官网显示,公司拥有5条管线。对于一家未上市的biotech来说,这可谓挑战巨大。2022年3月,在镇定思痛之后,Bone Biologics决定彻底改革整个产品线,将所有开发资源,集中于高风险胫骨骨折的异基因细胞疗法。随后,Bone基本上解雇了整个核心高管团队——CEO、CSO、CFO、CBO都被扫地出门,非执行董事会成员停职留薪。此前有披露称,如果Bone继续正常运营,它将在今年第三季度耗尽资金。幸运的是,Bone被法国Biotech Medsenic拯救,在8月份对其进行了反向合并。两家公司合并后更名为BioSenic,并将致力于炎症和骨科疗法的研发。Bone的细胞疗法是BioSenic保留的资产之一,其IIb期试验结果将于2023年上半年公布。在这笔交易中,Bone的最终估值略高于1000万美元。合并,使Bone在2022年不会踏入墓地,但不代表以后不会。手里仅有510万美元库存现金,BioSenic还远谈不上安全。来源 | 氨基观察(药智网获取授权转载)撰稿 | 朱来责任编辑 | 八角声明:本文系药智网转载内容,图片、文字版权归原作者所有,转载目的在于传递更多信息,并不代表本平台观点。如涉及作品内容、版权和其它问题,请在本平台留言,我们将在第一时间删除。— Tips —如果您有医药行业相关的线索或对医药政策、研发创新、资本市场、行业发展有深度观察欢迎投稿给我们马老师 18323856316(同微信)邮箱:maxuelian@yaozh.com 阅读原文,是昨天最受欢迎的文章哦
IPO
100 项与 SCN9A Candidate 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 神经痛 | 临床1期 | 美国 | 2021-12-03 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

特殊审评
只需点击几下即可了解关键药物信息。
登录
或

生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用