预约演示
更新于:2026-05-20
Zhejiang Hengyu Biotechnology Co., Ltd.
更新于:2026-05-20
概览
关联
100 项与 浙江恒驭生物科技有限公司 相关的临床结果
登录后查看更多信息
0 项与 浙江恒驭生物科技有限公司 相关的专利(医药)
登录后查看更多信息
97
项与 浙江恒驭生物科技有限公司 相关的新闻(医药)2026-05-18
一
什么是支原体?
支原体(Mycoplasma)是一类介于细菌与病毒之间、无细胞壁的原核细胞型微生物。
1898 年,法国学者 Nocard 与 Roux 首次从牛肺疫病灶中分离出支原体,当时命名为类胸膜肺炎微生物(Pleuropneumonia-like organism, PPLO)。
1967 年,国际分类学委员会正式将类胸膜肺炎微生物(Pleuropneumonia-like organism, PPLO)定名为支原体(Mycoplasma)。
截至目前,全球已发现并分离的支原体超过 240 种。
二
为何生物制药高度重视支原体防控?
不同支原体的致病机制存在差异,主要可分为三类:
1、 支原体对宿主细胞的直接损伤
支原体自身几乎无法合成氨基酸、脂肪酸、辅助因子及维生素,需与宿主细胞竞争上述前体物质,直接导致宿主细胞功能受损。
支原体通过黏附素(adhesin)结构黏附于宿主细胞表面,干扰细胞膜受体功能,引发细胞损伤。
因支原体无细胞壁,易与宿主细胞膜发生融合,其胞内酶系可干扰宿主细胞正常代谢。
部分支原体(如猪滑液支原体 M. hyosynoviae)可引发显著细胞病变,直接诱导细胞凋亡与坏死。
2、 支原体感染诱发的免疫病理损伤
如肺炎支原体(M.pneumoniae)感染可引发溶血、神经系统损伤、肾小球肾炎等并发症。
3、 部分支原体存在潜在致癌风险
如猪鼻支原体膜脂蛋白 p37 可结合人 EGFR 与 ANXA2,激活 MAPK、Akt、NF-κB 等信号通路,促进胃癌转移;p37 亦可通过与 HER2 相互作用促进其磷酸化,激活 ERK1/2 通路,加速乳腺肿瘤进展。
三
支原体检测相关法规要求?
ICH Q5D:《Quality of Biotechnological Products: Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biological Products》:要求对MCB/WCB/EOPC、种子批、收获液、终产品进行支原体检测。
《中国药典》0234 生物制品生产用动物细胞基质制备及质量控制:要求MCB/WCB/ 生产终末细胞(EOPC)每代次必检支原体;使用昆虫细胞或植物源性材料时,新增螺原体、中间原体属、虫原体属检测。
FDA《Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals(1993)》:要求对细胞库、种子批、未加工收获液、终产品进行全流程支原体检测。
药典标准检测方法:
《中国药典》3301 支原体检查法
《欧洲药典》2.6.7 Mycoplamas
《美国药典》63 Mycoplasma Tests
《美国药典》77 Mycoplasma NAT
《日本药典》Mycoplasma Testing for Cell Substrates used for the Production of Biotechnological/Biological Products
四
主流支原体检测方法优缺点对比
检测方法
检测时长
核心优缺点
培养法
28 天
- 支原体检测金标准,方法成熟; - 灵敏度高; - 检测周期极长; - 需加入阳性菌株质控,存在交叉污染风险。
指示细胞培养法
约21 天
- 较培养法耗时略短,但仍无法满足细胞治疗等快检需求; - 灵敏度偏低; - 阳性质控引入污染风险。
NAT 法
约1 天
- 检测快速; - 无需阳性菌株,大幅降低污染风险; - 灵敏度高; - 需针对支原体多样性完成完整方法学验证。
五
支原体经典培养检测方法原理
1、支原体直接培养法
支原体直接培养法采用液体培养基与固体 / 半流体培养基,在提供碳源、氮源及促生长因子的基础上,添加酚红作为 pH 指示剂。通常情况下,发酵型支原体产酸使培养基 pH 下降,颜色变黄;精氨酸分解型支原体产碱使 pH 上升,颜色变红。通过培养基颜色变化判断支原体生长,并观察固体 / 半流体培养基上的特征菌落:固体培养基可见 “荷包蛋” 样菌落,半流体培养基可见 “彗星状” 菌落 ,以此判定是否存在支原体污染。
2、支原体指示细胞培养法
将供试品接种于指示细胞中培养后,用核酸荧光染料染色。根据观察到的染色结果确定是否存在支原体污染。阴性结果仅细胞核显荧光;阳性结果可见细胞核荧光 + 核外荧光,据此判断支原体存在。
六
支原体 NAT 检测技术的应用与发展
随着分子生物学技术进步,核酸检测技术(NAT) 已成为支原体快速检测的主流方向。目前 EP、JP、USP 均已发布专章对支原体 NAT 方法提出明确要求与规范。
伴随基因治疗、细胞治疗等短货架期生物制品快速发展,NAT 方法将成为支原体检测的核心技术。
基于 qPCR 的支原体快速检测已实现成熟商业化应用。未来技术发展将聚焦更快速度、更便捷操作、更高通量。如利用微流控、巢式PCR、数字PCR、宏基因组NGS等技术,也逐步成为支原体快速检测的有效手段。
七
法规对支原体替代方法(NAT)的验证要求
法规趋势:
1、提升 NAT 技术地位,鼓励药企使用 NAT 方法。
2、以风险评估为基础开展替代方法验证,阳参选择与生产引入风险匹配。
3、样品的代表性,兼顾胞内支原体,同步检测细胞与上清液。
4、针对检测体系的检测限,可以通过对支原体的富集、培养扩增等手段,增强检测系统的检出能力。
5、相较早期法规的要求,对替代方法使用的支原体阳参GC/CFU比值提出来更加具体的要求(GC/CFU小于10)
八
恒驭推出的支原体试剂盒
恒驭生物支原体 DNA 检测试剂盒采用荧光探针 qPCR 技术,针对支原体保守基因区域设计特异性引物探针,可定性检测 200+ 种支原体。试剂盒特异性优异,与革兰氏阳性菌、梭菌、链球菌等近缘细菌及 CHO、VERO、HEK293 等工程细胞无交叉反应;按照 EP 2.6.7、JP XVIII、USP〈77〉要求完成方法学验证,检测限 <10 CFU/mL,满足 NAT 替代培养法的国际药典灵敏度要求。
产品信息
■检测类型:支原体定性检测
■适用样本:主细胞库、工作细胞库、病毒种子批、细胞培养上清 / 细胞悬液及各类生物制品
■试剂盒规格:50 reactions/盒
■检测时间:2-3h
■检测限:<10 CFU/mL
■试剂盒效期:24 个月(规定条件下储存)
■运输和储存条件:-20℃ 长期保存;运输采用干冰 / 冰袋冷链
产品优势
■操作简便:样本制备和检测总时长<3h,无需28天培养法
■覆盖度广:可检测 200+ 种支原体
■特异性强:验证了多个工程细胞、样本基质和近缘物种等,均无交叉反应
■结果可靠:内置内参(IC)+ 阴 / 阳性质控(PC、NCS、NTC),有效避免假阴性 / 假阳性
如需更多产品性能信息,请邮件联系 products@hyqure.com !
或扫描二维码,联系小驭客服!
产品信息
货号
产品名称
规格
HYM002
支原体DNA检测试剂盒(PCR-荧光探针法)
50T
关于上海恒驭
上海恒驭生物科技有限公司隶属于浙江恒驭生物科技有限公司,我们是一家专注于生物制品质量控制检测产品的研发、生产和销售服务的供应商。恒驭生物以国际合规标准为依托,凭借卓越的质量和技术实力,打造专注于大分子药物和CGT药物的Biosafety一站式服务平台,通过了欧盟QP、CNAS、ISO9001审计认证,满足NMPA、FDA、EMA等全球监管机构的严格要求,为创新药企提供可靠的保障。恒驭生物累计服务400多家生物药企,项目及申报实战经验丰富,已经为150+生物药的申报案例及60+BLA和商业化项目提供关键性支持(其中65+海外申报项目),包括病毒清除验证、细胞库检定、商业化批放行、复制型病毒(RCL、rcAAV、RCR等)检测、放行检测试剂盒等服务。
试剂盒作为恒驭生物的主营产品,严格符合中美等国家和地区相关的药品管理法规和标准,包括但不限于《美国药典》(USP)、《欧洲药典》(Ph.Eur.)、《中国药典》(ChP)等,确保客户申报和放行检测满足监管要求。凭借母公司强大的技术服务能力和超400家客户资源,恒驭生物迅速拓展试剂盒产品线,为客户提供各类残留物检测(CHO、E.coli、HEK293等细胞残留物)、复制型病毒检测(RCL、RCR等)、安全性检测试剂盒以及微生物检测试剂盒。
2026-05-14
·恒驭生物
在治疗用重组蛋白药物的研发过程中,细胞库作为生产体系的起始环节,是质量控制体系的重要基础。其相关研究与检定资料,构成了药学申报资料的核心组成部分。
2026年4月,国家药监局药品审评中心发布《治疗用重组蛋白药物首次申报临床试验药学资料撰写指导原则》,在 ICH M4Q (R1) 框架下,对 IND 阶段药学研究资料提出了进一步细化的技术要求。其中,涉及细胞库(MCB、WCB、EOPC)及未加工收获液(UPB)的相关内容,主要集中于以下两个模块:
3.2.S.2.3 物料控制
3.2.A.2 外源因子的安全性评价
基于上述指导原则及 ICH 相关技术要求,本文对治疗用重组蛋白药物 IND 阶段细胞库检定要点及未加工收获液外源因子检测要求进行梳理与归纳。
一
3.2.S.2.3 物料控制——上游构建与细胞库及物料控制要点
该部分主要涵盖目的基因构建、表达载体设计、宿主系统建立、细胞库构建与检定,以及生产用原材料控制等内容,需在申报资料中进行系统性描述。
1.上游构建相关内容及要求
目的基因:应提供目的基因来源、改构位点及原因等信息,提供目的基因序列及氨基酸序列信息,详述目的基因筛选过程。
表达载体:详述表达载体构建及筛选过程,提供表达载体功能元件、目的基因序列确证结果及确证报告(如有)。
宿主细胞/宿主菌:明确宿主菌的表型和基因型,提供宿主细胞/宿主菌来源及其历史信息,包括传代培养以及改造过程、建库检验信息(如有)等。
2.工程细胞及细胞库构建
工程细胞/工程菌:详述工程细胞/工程菌构建及筛选过程,包括基因转导/转染方法、筛选条件、单克隆挑选、优选克隆筛选等。
细胞库/菌种库:详述细胞库/菌种库建立过程以及检定结果,包括细胞库/菌种库名称、批号、代次、培养基组分、冻存液组分、传代条件、冻存条件、冻存细胞密度、保存条件等信息,并以附件形式提供细胞库/菌种库检定报告。详述细胞库/菌种库稳定性研究过程及结果(如有)。
3.生产用原材料控制
原材料基础信息:列表提供培养基、添加物、试剂、层析介质、滤膜等原材料的生产商、药典标准符合情况、使用阶段等信息;提供主要原材料内控标准(如有),关键原材料附COA文件。
人源/动物源性材料:明确建库及生产中是否使用人源/动物源性材料;如有,说明使用阶段、使用量,并进行安全性风险评估。提供TSE/BSE声明或供应商外源因子控制等证明文件。
自制原材料:明确是否使用人源/动物源性材料;提供上游构建、生产工艺、质量研究与控制、稳定性研究等资料。若采用CHO等真核细胞表达生产,工艺中应包含病毒去除/灭活单元,提供病毒清除验证研究方案及结果,并附验证报告。
二
细胞库检定项目及相关要求
主细胞库(MCB)及工作细胞库(WCB)通常在 GMP 条件下建立;生产终末细胞(EOPC,如适用)一般来源于临床批次制备。细胞库检定应采用符合 ICH 指南/药典要求的方法,并提供完整的原始检定报告。
以下为信息列表示例格式结果如下:
三
细胞库稳定性研究(如有)
依据《中国药典》2025年版 三部 0234生物制品生产用动物细胞基质制备及质量控制:细胞库稳定性研究包括传代稳定性研究和贮存稳定性研究(如有)。
传代稳定性研究:在规定传代水平(PDL)下,评估细胞生长状态、细胞活力、表达量、产品质量(活性、纯度、电荷变异体、糖基化、肽图等)、遗传稳定性(基因拷贝数、目的基因序列等)。各代次目的基因序列应与理论序列一致,各考察项目无显著差异。根据结果拟定WCB限传代次,可满足拟定生产规模的需求。
贮存稳定性研究:根据实际研究情况描述,可参考传代稳定性研究格式。
四
3.2.A.2 外源因子安全性评价——未加工收获液(UPB)检测要点
外源因子安全性评价需提供与外源因子有关的潜在污染风险评估信息,包括非病毒性外源因子及病毒性外源因子。从细胞系、动物和人源性材料、未加工收获液、病毒清除研究方面详述对病毒性外源因子的控制。
1.未加工收获液(UPB)资料与检测要求
未加工收获液(UPB),作为工艺早期关键中间产物,是评估污染风险、验证控制策略的重要“窗口”。
法规明确要求完整提供未加工收获液相关信息,包含样品批号、制备工艺过程、检验机构(第三方 / 自行检定)、检验项目、检验结果,可采用列表形式规范汇总,并以附件形式附 UPB 完整检定报告,确保数据可追溯、资料闭环。
UPB检测项目通常包括支原体、微生物限度、鼠细小病毒、内外源病毒污染检查等。检测方法及可接受标准应参照《中国药典》相应版本。以下为信息列表示例格式:对临床批(批号)UPB进行检测,提供了原始检测报告。
2.病毒清除验证合规要求(如适用)
应提供病毒清除验证研究过程及结果,以附件形式提供验证报告,包括如下:
验证机构;
模型病毒;
验证用样品(批号、工艺版本);
运行次数(示例中为2次);
缩小模型代表性分析;
验证结果及病毒安全性评估。
研究条件对比:应对比临床样品工艺参数范围与缩小模型验证工艺参数范围,明确经评估的最差工艺条件。例如低pH灭活步骤的温度、蛋白浓度、灭活pH、孵育时间等;病毒截留过滤步骤的滤器型号、跨膜压差(明确供应商验证的最差条件)、体积载量等。
验证研究结果汇总:以表格形式列出各工艺步骤、模型病毒及Log10下降因子(运行1、运行2)。
样品代表性评估:如未使用临床申报工艺代表性批次样品进行病毒清除验证,且验证用样品所用工艺与临床样品工艺间存在变更,应提供各验证步骤上样样品的质量对比,包括但不限于蛋白浓度、杂质水平等。
平台工艺:如涉及平台工艺,可参照《治疗用重组蛋白产品临床试验申请病毒清除工艺平台验证技术指导原则(试行)》提供相应的研究依据。
3.安全因子计算
按照最大临床剂量以及UPB中最大逆转录病毒样颗粒数量等最差条件进行计算,得出每剂量中可能存在的潜在逆转录病毒颗粒数量(示例:每XXX剂量中可能会有一个潜在的逆转录病毒颗粒)。该计算用于支持病毒安全性评价结论。
五
结语
IND 申报阶段中,细胞库检定、稳定性评价及外源因子控制相关资料,是药学申报体系的关键内容,也是评价产品安全属性、管控外源污染风险、维持质量稳定的重要技术支撑。依据 CDE 最新指导原则系统完成各项研究与资料梳理,可从生产源头严控生物安全风险,充分保障临床试验受试者安全,为项目合规申报与临床推进奠定坚实基础。
作为专业生物医药质控服务平台,恒驭生物细胞库检定业务,专注为大分子药物及 CGT 领域客户,提供一站式、全项覆盖、全球合规的质量控制整体解决方案。服务全面覆盖细胞种子、PCB、MCB、WCB、EOPC、UPB 等全类型生物样品;检测能力包括细胞鉴别、纯度分析、生物安全性检测、复制型病毒(RCL、RCR、rcAAV 等)筛查、稳定性与单克隆源性分析、成瘤及致瘤性研究等全维度项目,所有检测方法均完成系统方法学验证,符合 ICH、WHO 及中美欧等各国药典与药品注册申报规范要求。
公司搭建行业领先、运行稳定的专业化检测平台,配备先进的软硬件条件,包括透射电子显微镜、无菌隔离器、梅里埃 BACT/ALERT 3D 快速无菌检测系统、核型分析仪等高端精密设备,为精准检测、稳定质控筑牢硬件基础。同时,恒驭生物建立并严格运行GxP 质量管理体系,获得欧盟QP GMP符合性声明、CNAS 认可、ISO9001 认证等多项权威资质;部署有符合FDA Part 11、欧盟GMP附录11以及GAMP5等要求的实验室信息管理系统与在线环境监测系统,遵循ALCOA + CCEA数据完整性原则,保障检测数据真实、完整、可追溯。公司出具的检测报告合规规范、支持全球互认,满足客户海内外多地申报的需求。
深耕细胞库检定领域多年,恒驭生物积淀了深厚的海内外注册申报经验,行业服务口碑突出:已累计服务 400 余家国内外创新药企,顺利支撑 150 余项新药申报项目、60 余项 BLA 生物制品许可申请及商业化落地项目,其中海外申报案例超 65 项,业务覆盖单抗、双抗、ADC、疫苗、重组蛋白、CAR-T、基因治疗、干细胞等大分子药物与细胞基因治疗全赛道。
凭借完备的检测能力、严苛的质量管控体系与丰富的全球申报实战经验,恒驭生物全程赋能创新药研发全周期,护航客户实现从早期研发、临床申报到商业化生产的高效合规落地,以专业质控实力为全球创新药产业高质量发展持续赋能。
关于恒驭生物
恒驭生物以国际合规标准为依托,凭借卓越的质量和技术实力,打造专注于大分子药物和 CGT 药物的 Biosafety 一站式服务平台,通过了欧盟 QP、CNAS、ISO9001 审计认证,满足 NMPA、FDA、EMA 等全球监管机构的严格要求,为创新药企提供可靠的保障。恒驭生物累计服务400多家生物药企,项目及申报实战经验丰富,已经为150+生物药的申报案例及60+BLA和商业化项目提供关键性支持(其中65+海外申报案例),包括病毒清除验证、细胞库检定、商业化批放行、复制型病毒(RCL、rcAAV、RCR等)检测、HCD与HCP试剂盒与检测等服务。
恒驭生物拥有实战经验丰富的技术及质量团队,采用先进的系统和管理体系,包括符合 ISPE GAMP5 管理要求的 LIMS、EMS 系统,建立了遵循 ALCOA+ 原则的数据管理体系。无论是生物制品的 IND、BLA 申报还是商业化生产的全生命周期管理,恒驭都能够提供可靠的生物安全和质量控制支持,为客户的成功提供有力保障。
2026-05-13
·恒驭生物
2026 In Vivo X 创新峰会将于 2026 年 5 月 19-20 日在上海张江科学城希尔顿酒店正式举办。本次大会以 “Accelerating ideas, Delivering impact”为主题,汇聚超过50位重磅嘉宾,800+行业精英参会,大会由主论坛+5大专题论坛+BD项目路演专场组成,精心打造集技术交流、产业对接于一体的In vivo行业平台。
恒驭生物研发总监胡孟军博士,受邀将于5月20日上午在质量与法规前沿分论坛,做专题演讲:体内细胞治疗产品申报的生物安全性检测策略的,届时诚邀各位参会同仁莅临交流探讨符合国际申报要求的病毒清除工艺验证、细胞库检定、复制型病毒检测(RCL、RCR、rcAAV、RCA)、工艺残留物检测等优质服务。
5月20日上午专题演讲
胡孟军博士
同济大学博士毕业,曾就职于吉凯基因、上海齐鲁制药研究中心,拥有8年以上的生物药质量分析经验,参与过多个已进入临床阶段的抗体药物早期研发阶段的成药性评价。现任恒驭生物研发总监,建立了干细胞检测、病毒载体表征分析、复制型病毒检测、试剂盒开发、NGS测序等平台。
《体内细胞治疗产品申报的生物安全性检测策略》
生物安全检测的必要性
法规要求及审评关注点
生物安全检测策略
届时,欢迎广大客户及专家莅临现场,交流中美欧等多地申报经验,共话创新药生物安全与申报上市。
会议基本信息
会议时间:2026年5月19-20日
会议地点:上海市张江科学城希尔顿酒店3F
演讲论坛:分论坛四:质量与法规前沿
嘉宾及日程概览
重磅嘉宾
上下滑动查看更多嘉宾
会议日程
上下滑动查看完整议程
关于恒驭生物
恒驭生物以国际合规标准为依托,凭借卓越的质量和技术实力,打造专注于大分子药物和 CGT 药物的 Biosafety 一站式服务平台,通过了欧盟 QP、CNAS、ISO9001 审计认证,满足 NMPA、FDA、EMA 等全球监管机构的严格要求,为创新药企提供可靠的保障。恒驭生物累计服务400多家生物药企,项目及申报实战经验丰富,已经为150+生物药的申报案例及60+BLA和商业化项目提供关键性支持(其中65+海外申报案例),包括病毒清除验证、细胞库检定、商业化批放行、复制型病毒(RCL、rcAAV、RCR等)检测、HCD与HCP试剂盒与检测等服务。
恒驭生物拥有实战经验丰富的技术及质量团队,采用先进的系统和管理体系,包括符合 ISPE GAMP5 管理要求的 LIMS、EMS 系统,建立了遵循 ALCOA+ 原则的数据管理体系。无论是生物制品的 IND、BLA 申报还是商业化生产的全生命周期管理,恒驭都能够提供可靠的生物安全和质量控制支持,为客户的成功提供有力保障。
临床申请细胞疗法一致性评价
100 项与 浙江恒驭生物科技有限公司 相关的药物交易
登录后查看更多信息
100 项与 浙江恒驭生物科技有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月08日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
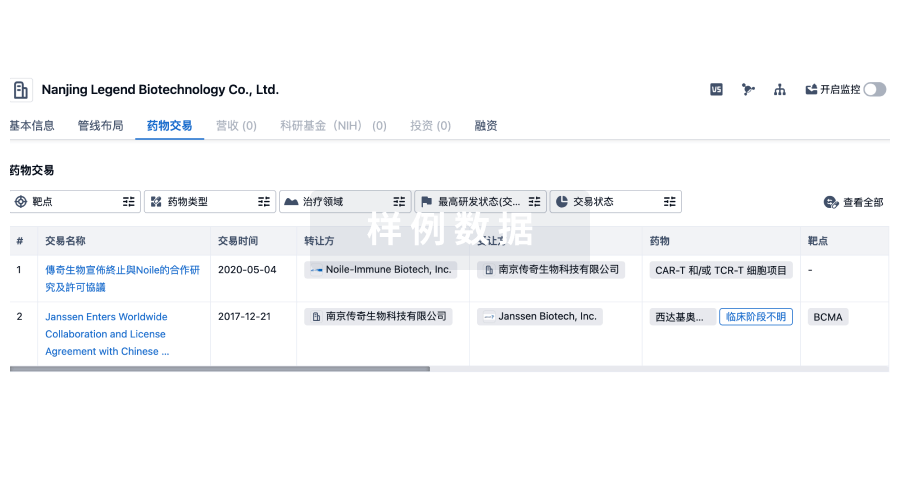
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
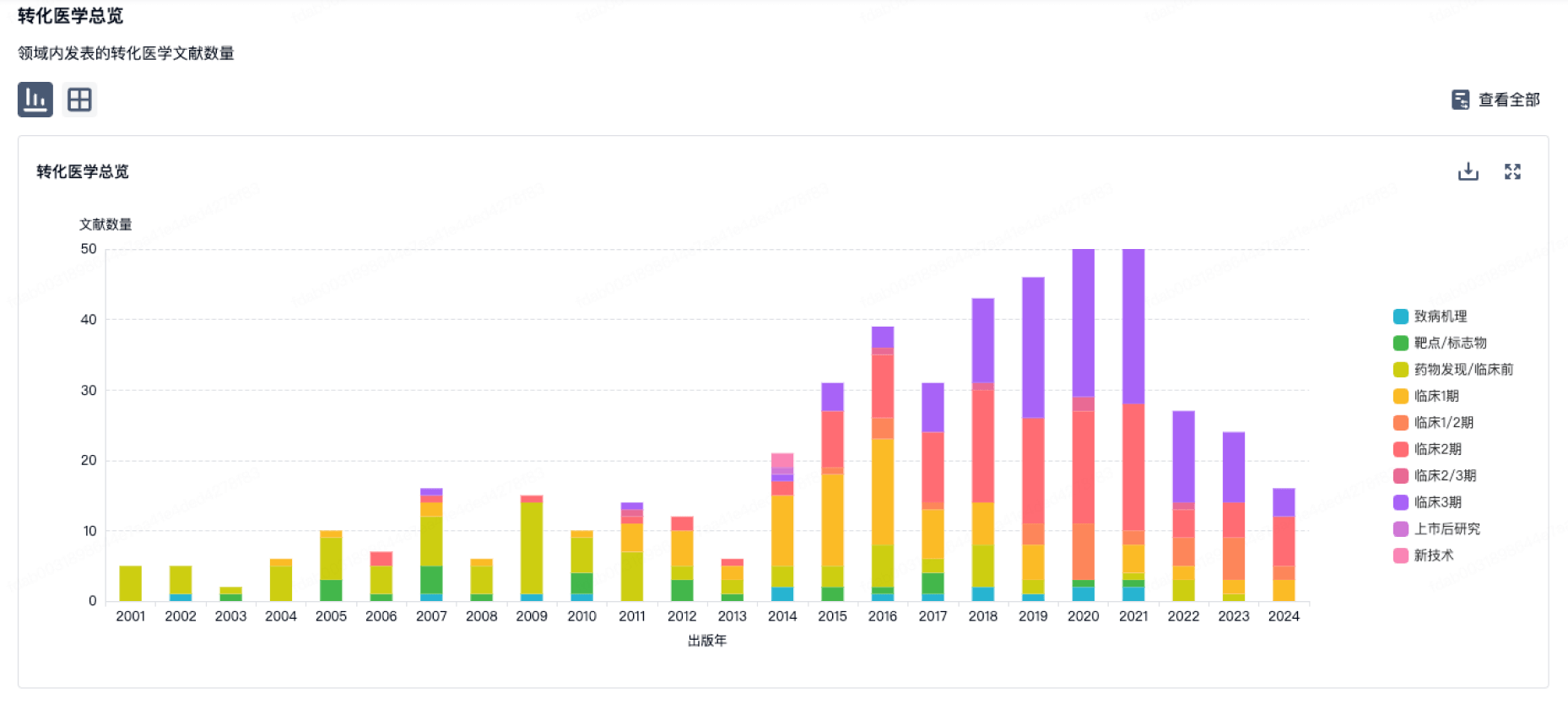
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
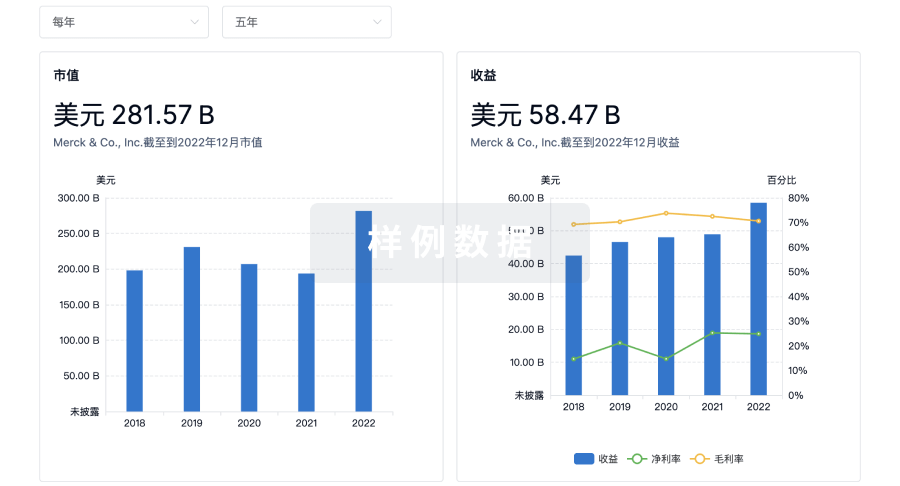
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用