预约演示
更新于:2026-03-05

Takara Bio USA, Inc.
更新于:2026-03-05
概览
关联
100 项与 Takara Bio USA, Inc. 相关的临床结果
登录后查看更多信息
0 项与 Takara Bio USA, Inc. 相关的专利(医药)
登录后查看更多信息
86
项与 Takara Bio USA, Inc. 相关的文献(医药)2025-04-09·Precision Clinical Medicine
A benchmarking study of copy number variation inference methods using single-cell RNA-sequencing data
Article
作者: Zhang, Xiaowen ; Xiao, Chunlin ; Farmer, Andrew ; Mason, Christopher E ; Wu, Shixiu ; Zhu, Bin ; Xiong, Wei ; Moos, Malcolm ; Wu, Hongjin ; Jones, Wendell ; Wang, Charles ; Gong, Shusheng ; Chen, Wanqiu ; Chen, Xin ; Chen, Zhong ; Fang, Li Tai
Abstract:
Background:
Single-cell RNA-sequencing (scRNA-seq) has emerged as a powerful tool for cancer research, enabling in-depth characterization of tumor heterogeneity at the single-cell level. Recently, several scRNA-seq copy number variation (scCNV) inference methods have been developed, expanding the application of scRNA-seq to study genetic heterogeneity in cancer using transcriptomic data. However, the fidelity of these methods has not been investigated systematically.
Methods:
We benchmarked five commonly used scCNV inference methods: HoneyBADGER, CopyKAT, CaSpER, inferCNV, and sciCNV. We evaluated their performance across four different scRNA-seq platforms using data from our previous multicenter study. We evaluated scCNV performance further using scRNA-seq datasets derived from mixed samples consisting of five human lung adenocarcinoma cell lines and also sequenced tissues from a small cell lung cancer patient and used the data to validate our findings with a clinical scRNA-seq dataset.
Results:
We found that the sensitivity and specificity of the five scCNV inference methods varied, depending on the selection of reference data, sequencing depth, and read length. CopyKAT and CaSpER outperformed other methods overall, while inferCNV, sciCNV, and CopyKAT performed better than other methods in subclone identification. We found that batch effects significantly affected the performance of subclone identification in mixed datasets in most methods we tested.
Conclusion:
Our benchmarking study revealed the strengths and weaknesses of each of these scCNV inference methods and provided guidance for selecting the optimal CNV inference method using scRNA-seq data.
2021-12-15·Journal of biomolecular techniques : JBT
Benchmarking Single-Cell mRNA–Sequencing Technologies Uncovers Differences in
Sensitivity and Reproducibility in Cell Types With Low RNA Content
Article
作者: Melloy, Leigh Ann ; Farmer, Andrew ; Duong, Tommy ; Bansal, Nidhanjali ; Holcomb, Ilona ; Laliberte, Julie ; Hildebrand, Jerry ; Swaminathan, Karthikeyan ; Xu, Hui Helen ; Babb, Paul
Single-cell RNA sequencing (scRNA-seq) has the ability to classify each cell and determine the transcriptomic profile of specific cell types and cells of a given disease state; however, sensitivity of the gene count for each cell can be a critical component to the success of a single-cell study. The recently introduced SMART-Seq Single Cell PLUS Kit (SSsc PLUS) claims to provide higher sensitivity and reproducibility versus popular methods for the sequencing analysis of single cells. Here, the cDNA-generation component of the kit, SMART-Seq Single Cell Kit (SSsc), was compared with the popular homebrew protocol, Smart-seq2, and its update, Smart-seq3. The SMART-Seq Library Prep Kit from SSsc PLUS was benchmarked against a commonly used scRNA-seq library preparation method, Illumina Nextera XT. Finally, the SSsc chemistry was tested in both full and fractional volumes on 2 popular liquid-handler devices to investigate whether the high sensitivity was maintained in miniaturization. We demonstrate that SSsc PLUS outperforms these other full-length methods in convenience, sensitivity, gene identification, and reproducibility while also offering full compatibility with automation platforms.
2021-07-21·Nucleic acids research2区 · 生物学
Measuring nonhomologous end-joining, homologous recombination and alternative end-joining simultaneously at an endogenous locus in any transfectable human cell
2区 · 生物学
ArticleOA
作者: Viale, Agnes ; Powell, Simon N ; Hussain, Suleman S ; Rong-Mullins, Xiaoqing ; Leeman, Jonathan E ; Sifuentes, Christopher ; Higginson, Daniel S ; Majumdar, Rahul ; Anderson, Kyrie S ; Mohibullah, Neeman ; Soni, Rekha ; Farina, Andrea ; Hamzic, Edin ; Damerla, Rama R ; Li, Yi ; Ravindran, Pavithran T ; Jalan, Manisha ; Bazil, Maximilian J ; Moore, Grace M ; Buechelmaier, Erika S ; Narang, Himanshi
Abstract:
Double strand break (DSB) repair primarily occurs through 3 pathways: non-homologous end-joining (NHEJ), alternative end-joining (Alt-EJ), and homologous recombination (HR). Typical methods to measure pathway usage include integrated cassette reporter assays or visualization of DNA damage induced nuclear foci. It is now well understood that repair of Cas9-induced breaks also involves NHEJ, Alt-EJ, and HR pathways, providing a new format to measure pathway usage. Here, we have developed a simple Cas9-based system with validated repair outcomes that accurately represent each pathway and then converted it to a droplet digital PCR (ddPCR) readout, thus obviating the need for Next Generation Sequencing and bioinformatic analysis with the goal to make Cas9-based system accessible to more laboratories. The assay system has reproduced several important insights. First, absence of the key Alt-EJ factor Pol θ only abrogates ∼50% of total Alt-EJ. Second, single-strand templated repair (SSTR) requires BRCA1 and MRE11 activity, but not BRCA2, establishing that SSTR commonly used in genome editing is not conventional HR. Third, BRCA1 promotes Alt-EJ usage at two-ended DSBs in contrast to BRCA2. This assay can be used in any system, which permits Cas9 delivery and, importantly, allows rapid genotype-to-phenotype correlation in isogenic cell line pairs.
21
项与 Takara Bio USA, Inc. 相关的新闻(医药)2025-12-29
·生物学报
生物学报
撰文:王 萌
编辑:武钰婷
2025 年末,生物制药行业传来振奋人心的信号:曾在 2021-2024 年深陷熊市的生物科技板块强势反弹,SPDR S&P 生物科技 ETF 从 4 月每股 66.66 美元的低位一路飙升,12 月 19 日收于 123.43 美元,涨幅高达 85%。这一反弹背后,是特朗普政府制造业回流政策的落地、AI 技术在医药领域的深度渗透、基因疗法的突破性进展以及全球资本对生物制药赛道的重新聚焦。2026 年,将成为生物制药行业告别波动、迈向价值创造的关键转折年。以下七大核心趋势,将全面揭秘这个万亿赛道的新机遇与新变革。一、AI 制药:从概念炒作到价值兑现,贯穿新药研发全链条
人工智能在生物制药领域的应用,终于走过了 “纸上谈兵” 的阶段,2026 年将迎来规模化落地的 “价值兑现期”。德勤 2026 年生命科学展望调查显示,78% 的生物制药高管认为 AI 将成为驱动行业重大变革的核心力量,29% 的企业计划通过 AI 工具或相关培训提升员工生产力。不过,行业仍面临现实挑战:在更广泛的生命科学领域(含医疗技术),仅有 22% 的领导者表示成功实现了 AI 规模化应用,仅 9% 报告获得了显著的投资回报,14% 的高管表示已将 AI 工具全面融入日常工作流程,另有 40% 正在朝这一目标推进。
2026 年,AI 的发力点将集中在三大核心场景。首先是智能临床试验优化,AI 制药企业 Recursion 推出 “ClinTech” 计划,通过三大模块革新临床试验模式:智能试验设计让方案更精准,加速患者招募缩短启动周期,强化证据生成提升数据可信度,有望将新药研发周期缩短数月甚至数年。其核心候选药物 REC-1245 将于 2026 年上半年公布 I 期临床安全性和药代动力学数据,用于治疗生物标志物富集的实体瘤和淋巴瘤,同时还计划启动 REC-102 用于低磷酸酯酶症、REC-7735 用于 HR + 乳腺癌的 I 期试验。
其次是全流程效率提升,从药物分子设计、靶点筛选到生产工艺优化,AI 正贯穿医药研发的每一个环节。普华永道制药与生命科学交易负责人 Roel van den Akker 指出:“2026-2027 年的行业关键词是‘价值落地’,企业需要明确 AI 在临床开发和商业推广中到底能发挥什么实际作用。如果能通过 AI 缩短研发时间、提高商业化效率,这些价值都将直接体现在交易估值和运营业绩中。” 此外,AI 在并购尽职调查中的应用也将常态化,成为企业评估标的价值的重要工具。
AI 制药企业的资本化进程也在加速。2025 年 12 月底,AI 制药龙头英矽智能(Insilico Medicine)在香港交易所上市,计划发行 9469 万股,每股定价 24.05 港元,募资 20.26 亿港元(约合 26.05 亿美元),腾讯、礼来等 15 家行业巨头成为基石投资者。此次募资将主要用于药物研发、AI 平台升级和自动化实验室扩建。其核心药物 RENTOSERTIB(TNIK 抑制剂)将于 2026 年上半年在中国启动特发性肺纤维化的 IIb/III 期试验,同时向美国 FDA 提交吸入式 RENTOSERTIB 用于特发性肺纤维化、小分子 RENTOSERTIB 用于肾纤维化的临床试验申请。英矽智能还与礼来扩展了合作,有望从这家制药巨头获得超过 1 亿美元的合作收入。二、千亿资本回流美国:4800 亿打造本土生物制造帝国
为响应特朗普政府的 “制造业回流” 政策,规避海外生产可能面临的 100% 关税,全球生物制药巨头纷纷加大对美国本土的投资力度。据美国外交关系协会数据,截至 2025 年 11 月中旬,生物制药企业已承诺投资超过 4800 亿美元,用于在美国建设生产基地和研发中心。这一趋势不仅重塑了全球生物制药供应链格局,更让美国多个 “非传统生物产业州” 迎来了发展机遇。
在这场投资热潮中,辉瑞和默克(Merck & Co.)成为领跑者,两家企业各承诺投资 700 亿美元,合计占总投资额的近三分之一。2025 年 9 月,辉瑞 CEO Albert Bourla 在白宫公开承诺,将为美国医疗补助计划(Medicaid)患者提供最惠国定价(MFN),以此换取三年的关税豁免。尽管辉瑞尚未明确投资的具体落地地点,但这一巨额承诺彰显了企业对美国本土市场的重视。默克的投资计划则更为具体,将斥资 30 亿美元在弗吉尼亚州埃尔克顿建设制药生产卓越中心,专注于小分子药物生产,预计创造 500 多个全职永久岗位,该中心已于 2025 年 9 月破土动工,计划 2028 年投产,2030 年开始商业化生产。
弗吉尼亚州成为此次投资热潮的最大赢家之一。2025 年 10 月,阿斯利康宣布投资 45 亿美元在夏洛茨维尔附近的阿尔伯马尔县建设生产基地,计划创造 600 多个永久岗位,这是阿斯利康 500 亿美元美国投资计划的重要组成部分。该基地最初计划生产减肥药和代谢疾病药物,包括口服 GLP-1、巴克斯罗他汀、口服 PCSK9 抑制剂等,后来扩展至抗体偶联药物(ADC)等抗癌药物的生产。长期以来,弗吉尼亚州在生物健康领域一直位居马里兰州之后,此次凭借大额投资有望实现弯道超车,成为生物制造的新兴高地。
除了弗吉尼亚州,美国多个州都迎来了生物制药巨头的布局。阿拉巴马州将迎来礼来投资 60 亿美元建设的活性药物成分(API)生产厂,用于生产小分子药物和多肽类药物(如口服 GLP-1 受体激动剂奥福格列汀),礼来计划 2026 年底前为该药物提交上市申请,该工厂将创造 450 个永久岗位。印第安纳州,礼来正在建设投资 45 亿美元的 “医药铸造厂”,整合研发与生产功能,预计 2027 年底投产,创造 400 个就业岗位。俄亥俄州,安进将其哥伦布市附近工厂的投资从 9 亿美元提升至 14 亿美元,预计雇佣 750 人。得克萨斯州,礼来计划投资 65 亿美元在休斯顿建设 API 工厂,创造 615 个就业岗位。
CBRE 美国生命科学咨询业务负责人 Matt Gardner 表示:“这些投资并非简单的产能转移,而是在新区域构建完整的生物制造生态系统。这不仅为当地带来了就业和经济增长,也让生物制药行业的地理版图更加多元化,打破了传统生物产业集群的垄断格局。”三、癌症治疗革命:双抗挑战 “药王”,mRNA 疫苗多点突破
癌症治疗一直是生物制药行业的核心赛道,2026 年将迎来两大突破性方向:新型双特异性抗体向传统 “药王” 发起挑战,mRNA 疫苗从传染病领域跨界抗癌,为癌症患者带来新的治疗选择。
在靶向治疗领域,辉瑞与中国生物制药企业 3SBio 合作开发的 PF-08634404(原 SSGJ-707)成为焦点。这款下一代双特异性抗体同时靶向 PD-1 和 VEGF 两大靶点,有望成为癌症治疗的 “新基石”,尤其在疾病早期阶段,可能无需联合化疗即可达到显著疗效。2026 年,辉瑞计划启动两项 III 期临床试验,分别评估该药在局部晚期或转移性非小细胞肺癌、转移性结直肠癌中的疗效,同时开展肝癌、肾癌、膀胱癌的 I/II 期研究,探索与抗体药物偶联物 Pacdev(enfortumab vedotin-ejfv)的联合治疗方案。
这款药物的目标直指默克的 “药王” Keytruda(帕博利珠单抗)。Keytruda 作为一款多适应症癌症免疫疗法,2024 年销售额达 294.82 亿美元,2025 年前三季度销售额再创新高,达 233.03 亿美元,长期占据全球畅销药榜首。辉瑞在 2025 年 7 月与 3SBio 达成协议,获得该药除中国以外的全球权益,支付 12.5 亿美元预付款和 1 亿美元股权投资,若行使中国权益选择权,还将支付最高 1.5 亿美元。短短六个月内,辉瑞已完成三项临床试验申请,成功生产首批药物,并选定全球 500 多个临床试验站点,推进速度之快彰显了其对这款药物的信心。
mRNA 技术在癌症疫苗领域的应用也取得了重大进展。Moderna 与默克合作开发的个性化新抗原疫苗 intismeran autogene(原 mRNA-4157/V940),已完成黑色素瘤 III 期辅助治疗试验的入组,预计 2026 年公布关键疗效数据。该疫苗是一种个体化新抗原疗法(INT),与 Keytruda 联合使用,目前正在开展三项 III 期临床试验,除黑色素瘤外,还包括两项针对非小细胞肺癌的研究,均已启动患者招募。此外,该疫苗还在开展肾癌、膀胱癌、转移性黑色素瘤等多个适应症的 II 期试验,Moderna 计划公布另一项黑色素瘤辅助治疗 II 期试验的五年随访数据,进一步验证其长期疗效。
BioNTech 与再生元的合作也传来好消息。在 2025 年欧洲肿瘤内科学会(ESMO)大会上,双方公布了 mRNA 癌症疫苗 BNT111 联合 PD-1 抑制剂 Libtayo(cemiplimab)治疗 PD-(L) 1 耐药 / 复发的不可切除 III 期或 IV 期黑色素瘤的 II 期试验数据。结果显示,联合疗法的客观缓解率(ORR)达 18.1%,显著高于历史对照的 10%;24 个月时,37% 的患者仍存活,21% 的患者无肿瘤进展。BNT111 基于 BioNTech 的 FixVac 平台,编码四种黑色素瘤相关抗原,为耐药患者提供了新的治疗选择。四、投融资回暖:并购热潮涌动,IPO 市场有望复苏
经历数年的行业寒冬后,生物制药投融资市场在 2025 年率先回暖,2026 年将持续保持活跃态势。全球数据显示,2025 年生物制药并购交易额达 1796 亿美元,同比增长 31%,尽管交易数量从 2024 年的 583 宗下降至 468 宗,但大额交易成为市场主流,彰显了行业资源向优质资产集中的趋势。
专利悬崖是推动并购热潮的核心驱动力。根据 GEN 榜单,2026-2029 年全球即将失去专利保护的 TOP20 药物,2024 年合计销售额达 1764.42 亿美元,占同期即将流失专利销售额的 75%。为了填补管线空白,避免业绩下滑,制药巨头纷纷通过并购获取创新资产。2025 年,辉瑞在与诺和诺德的竞标中胜出,以最高 100 亿美元收购肥胖症药物开发商 Metsera;诺华宣布以 120 亿美元收购 Avidity Biosciences,交易预计 2026 年上半年完成;12 月 19 日,BioMarin Pharmaceutical 宣布以 48 亿美元收购 Amicus Therapeutics;Alkermes 则以最高 23.7 亿美元收购 Avadel Pharmaceuticals。
安永帕特农全球生命科学交易负责人 Subin Baral 表示:“除了专利悬崖压力,制药巨头的巨额现金储备也是并购活跃的重要原因。全球 TOP25 生物制药企业的现金储备高达 1.4 万亿美元,为并购提供了充足的资金支持。而许多创新企业由于 COVID-19 时期的风险投资逐渐耗尽,投资者态度趋于谨慎,现金流紧张,成为理想的并购标的。” 这些被并购的资产大多已进入中后期研发阶段,部分已实现商业化,降低了并购后的研发风险。
风险投资(VC)市场也呈现出回暖迹象,但投资结构发生了明显变化。2025 年前三季度,生物科技 VC 投资达 171 亿美元,涉及 290 宗交易,虽低于 2021 年的 444 亿美元(776 宗交易)和 2024 年的 272 亿美元(459 宗交易),但投资向后期阶段集中,大额融资增多。业内人士表示,尽管早期项目融资仍显不足,但行业基本面持续向好,后期项目的融资活跃为行业发展提供了重要支撑。
IPO 市场有望在 2026 年迎来复苏。2025 年仅有 11 家生物制药企业完成 IPO,远低于 2024 年的 24 家和 2021 年的峰值,IPO 融资额也从 2021 年的 227 亿美元降至 2025 年前三季度的 18 亿美元。不过,随着并购和 VC 市场的回暖,作为资本退出重要渠道的 IPO 市场正逐步积蓄力量。Truist Securities 生物制药投资银行负责人 Michael Allwin 表示:“市场需要一定的确定性才能恢复信心,2025 年后续融资活动(如增发、二次发售、PIPE 交易、可转换债券融资)的活跃是积极信号。IPO 通常是最后复苏的环节,我们预计 2026 年 IPO 市场将恢复至 2019 年的正常水平,虽难以重现 2020-2021 年的峰值,但将回归常态化。”五、基因疗法:从个案治愈到监管创新,开启个性化治疗时代
2025 年,基因疗法领域迎来了里程碑式的突破:17 个月大的 KJ Muldoon 成为全球首个通过个性化 CRISPR 疗法治愈罕见病的患者。KJ 患有严重的氨甲酰磷酸合成酶 1(CPS1)缺乏症,这是一种致命的遗传性代谢疾病。在费城儿童医院和宾夕法尼亚大学研究团队的合作下,通过个性化 CRISPR 疗法直接修正了其基因组中的致病突变,成功挽救了他的生命。这一案例被发表在《新英格兰医学杂志》上,45 位作者共同预测:“针对多种遗传疾病的患者特异性基因编辑疗法将快速普及,成为常规治疗手段。”
KJ 的成功治疗离不开多方协作,包括美国 FDA、加州大学伯克利分校创新基因组学研究所、马萨诸塞州总医院布莱根基因与细胞治疗研究所、丹纳赫集团和 Acuitas Therapeutics 等机构的深度参与。尤为值得关注的是,FDA 在收到治疗申请后仅用一周时间就完成了审批,为患者争取了宝贵的治疗时间。创新基因组学研究所技术与转化 director Fyodor Urnov 表示:“我们有能力为像 KJ 这样的美国儿童创造美好的未来,关键是要相信本土的创新解决方案。”
这一突破性案例也推动了监管政策的创新。2025 年 11 月,FDA 局长 Martin A. Makary 和生物制品评价与研究中心(CBER)主任 Vinayak Prasad 宣布推出 “合理机制路径”(PM Pathway),为超罕见病的个性化疗法开辟绿色通道。这类疾病患者数量极少,无法开展传统的随机对照临床试验,新路径允许基于科学合理的作用机制、临床前数据和初步临床结果加速审批,同时适用于缺乏有效治疗手段或现有治疗效果不佳的常见病。这一监管创新将极大地推动个性化基因疗法的发展,让更多罕见病患者受益。
不过,基因疗法的快速发展也伴随着安全性挑战。Sarepta Therapeutics 的 DMD(杜氏肌营养不良症)基因疗法 Elevidys 因三名患者死亡引发了行业广泛关注。2025 年 3 月,一名 16 岁男性患者在接受治疗后死于急性肝衰竭;6 月,第二名患者死亡;随后,一名 8 岁巴西男孩在治疗后死亡。FDA 一度要求 Sarepta 暂停所有 Elevidys 的销售,Sarepta 最初拒绝暂停可行走患者的用药,后于 7 月 21 日同意。几天后,巴西当局排除了该疗法与男孩死亡的关联,FDA 允许 Sarepta 恢复可行走患者的药物供应。这一事件凸显了基因疗法在安全性与可及性之间的平衡难题,也促使监管机构和企业更加重视临床试验的安全性监测。六、组学技术:空间生物学成新战场,多组学融合引领精准医疗
组学技术(包括基因组学、转录组学、蛋白质组学等)作为生物制药的 “底层工具”,2026 年将迎来技术升级与市场竞争的双重爆发。从单细胞分析到空间转录组学,技术突破正让研究人员能够更精准地解析疾病机制,为精准医疗提供更全面的数据支持。
空间生物学成为行业竞争的新焦点。Illumina 将于 2026 年商业化推出其在 2025 年 AGBT 大会上亮相的空间转录组技术,该技术可在单次实验中分析数百万个细胞的空间分布,捕获面积是现有技术的 9 倍,分辨率提升 4 倍,将极大地推动肿瘤微环境、神经科学等领域的研究。该技术将采用新的多模态分析平台,与 Illumina 的 NextSeq 和 NovaSeq 测序仪兼容。为了强化多组学布局,Illumina 在 2024 年收购了单细胞技术企业 Fluent BioSciences,计划在 2026 年完成多组学产品线的整合,实现基因组学、转录组学、蛋白质组学的一站式分析。
Takara Bio USA 也在空间生物学领域积极布局。2025 年 1 月,Takara 收购了空间基因组学先驱 Curio Bioscience,将其 Trekker 和 Seeker 两大空间生物学平台纳入麾下,与自身的单细胞基因组学工具形成互补。2025 年 10 月,Takara 推出了一系列产品更新,旨在让更多研究人员能够使用这一新兴技术,进一步扩大市场份额。
下一代测序(NGS)领域的竞争也日趋白热化。罗氏在 2025 年 AGBT 大会上推出了 Sequencing By Expansion(SBX)技术,该技术整合了罗氏此前从 Stratos Genomics 和 Genia Technologies 收购的技术,通过生化转化将 DNA 编码为 Xpandomer 分子(长度为目标 DNA 的 50 倍),并利用高信噪比的报告分子编码 DNA 序列信息,结合 CMOS 传感器模块,实现高准确度的单分子纳米孔测序。这一技术的推出,直接挑战了 Illumina 在 NGS 市场的垄断地位。
除了商业竞争,法律诉讼也成为市场竞争的重要手段。2025 年 10 月,罗氏测序解决方案部门联合 10x Genomics 起诉 Illumina,指控其侵犯了五项单细胞专利(三项归 10x 所有,两项由 10x 从罗氏诊断部门授权获得)。Illumina 表示 “强烈否认这些指控,并将积极辩护”。此外,10x 还联合 Prognosys Biosciences 针对四项空间生物学专利单独起诉 Illumina,这场法律与商业的双重较量将深刻影响测序市场的格局。七、政策环境趋稳:FDA 提速审批,产业政策持续利好
2025 年,特朗普政府的政策调整曾给生物制药行业带来一定的不确定性,但随着监管框架逐渐清晰,2026 年行业将迎来更稳定、更具可预测性的政策环境。
FDA 的改革成为行业关注的焦点。2025 年 4 月,特朗普提名的 Martin A. Makary 出任 FDA 局长后,提出了 “新 FDA” 计划,旨在通过 AI 和大数据提升药物审批效率,加强与行业的合作。2025 年 11 月,Makary 启动了 “ Commissioner’s National Priority Voucher” 试点项目,为符合条件的药物开发商提供快速审批通道:如果企业承诺提高药物可及性、在美国本土生产(作为国家安全问题)或解决未满足的公共卫生需求,其药物申请将在 1-2 个月内完成审核,远快于常规的 10-12 个月审批周期。这一政策将极大地激励企业聚焦关键治疗领域,同时推动制造业回流。
Truist Securities 的 Michael Allwin 表示:“Makary 领导下的 FDA 表现出了商业建设性,临床数据里程碑、试验终点设定等方面都回归了常态化,我们看到了更多符合预期的药物批准,审批节奏也更加稳定,这对行业发展是重大利好。” 尽管 2025 年 FDA 经历了 3500 人的裁员(后恢复四分之一岗位)和高层人事动荡,但目前监管方向已趋于稳定。
产业政策方面,最惠国定价(MFN)政策持续发挥作用。企业通过承诺为美国患者提供更低的药物价格,换取海外生产药物的关税豁免,这一政策既降低了患者用药成本,又推动了生物制药制造业向美国本土回流,实现了企业与患者的双赢。尽管美国药物定价问题仍存在一些未解决的争议,但行业普遍认为,2026 年的政策将更具可预测性,不会出现大幅波动。
基础研究投入方面,特朗普政府曾提议削减 NIH 40% 的预算,但这一提案遭到了国会拨款委员会的否决,NIH 此前实施的 15% 研究补助金间接成本上限政策也得到了美国最高法院的支持,为基础研究提供了稳定的资金保障。尽管 NIH 在 2025 年进行了 1200 人的裁员,但核心研究项目并未受到严重影响,为生物制药行业的长期创新奠定了基础。结语:2026,生物制药的价值回归与黄金时代
从 AI 技术的规模化落地到基因疗法的个性化突破,从千亿资本的本土布局到投融资市场的全面回暖,2026 年的生物制药行业正告别过去几年的波动与迷茫,进入以 “价值创造” 为核心的新阶段。威廉・布莱尔的分析师团队在报告中指出:“只要临床数据持续强劲,药物上市不受过度价格监管的阻碍,生物科技领域的专业投资将继续取得优异表现。”
对于普通患者而言,2026 年将有更多创新疗法上市,癌症、罕见病等疾病的治疗效果将得到显著提升,用药成本也可能进一步降低;对于投资者而言,并购、VC 和 IPO 市场的全面活跃,为把握万亿赛道机遇提供了多元化的渠道;对于行业本身而言,制造业回流带来的供应链稳定、技术创新带来的研发效率提升、政策优化带来的发展环境改善,将共同推动生物制药行业进入高质量发展的黄金时代。2026 年,让我们共同见证生物制药行业的蜕变与崛起,迎接一个更健康、更具创新活力的未来。
欢迎转发到朋友圈和微信群
扫描关注公众号+标星
(星标,不错过精彩推文)
加微信进专业交流群
(请备注单位及职务)
2025-06-16
DUBLIN--(BUSINESS WIRE)--The "Antimicrobial Resistance Diagnostic Markets, Strategies and Trends by Pathogen and Technology, With Executive Guides and Customization" has been added to ResearchAndMarkets.com's offering.
As antimicrobial resistance (AMR) continues to pose a significant threat to global health, innovative diagnostic technologies are taking center stage in the effort to mitigate this challenge. These advancements could potentially address the resistance dilemma even in the face of delayed development of new antibiotics. Cutting-edge diagnostic tools, surpassing traditional gene sequencing methods, are being researched and developed to seize the expanding market opportunities within this vital healthcare domain.
In an extensive analysis, the publisher sheds light on the technological landscape, covering a variety of sophisticated approaches for diagnosing pathogens and infections. We have pinpointed six major pathogen-related opportunities, underscoring their importance in the diagnosis and treatment of infectious diseases. Furthermore, we offer detailed market forecasts to aid stakeholders in navigating this rapidly evolving field.
The study profiles over 30 leading companies, from industry giants to emerging innovators, all dedicated to advancing diagnostic solutions in the face of AMR. These entities are leveraging new technologies to enhance the accuracy, efficiency, and speed of pathogen detection, which is crucial in managing infections and curbing the spread of resistance.
With AMR limiting the efficacy of current antibiotics, the role of diagnostics becomes even more critical. Enhanced diagnostic tools not only aid in identifying appropriate treatments faster but also enable more precise use of antibiotics, thereby contributing to the global effort to fight resistance.
Current and future diagnostic technologies promise healthcare professionals more robust options in the ongoing battle against AMR. By expanding the avenues for early detection and precise pathogen identification, these technologies are essential in managing infectious disease threats and influencing treatment outcomes.
As the world continues to grapple with AMR and its implications on public health, the development and implementation of advanced diagnostic solutions stand as a beacon of hope. The strategic insights and comprehensive market forecasts provided in the publisher's study serve as valuable resources for stakeholders aiming to capitalize on the innovations reshaping the fight against infectious diseases.
Key Topics Covered:
1 Market Guides
1.1 Antimicrobial Resistance - Strategic Situation Analysis
1.2 Guide for Executives and Business Development Staff
1.3 Guide for Management Consultants and Investment Advisors
2 Introduction and Market Definition
2.1 The Threat and Opportunity of Antimicrobial Resistance
2.2 Defining the Opportunity
2.2.1 Revenue Market Size
2.3 Methods and Sources
2.3.1 Methodology
2.3.2 Sources
2.3.3 Authors
2.4 Perspective: Healthcare and the IVD Industry
2.4.1 Global Healthcare Spending
2.5 Spending on Diagnostics
2.5.1 Important Role of Insurance for Diagnostics
3 Overview of a Dynamic Market
3.1 Players in a Dynamic Market
3.1.1 Diagnostic Test Developer
3.1.2 Instrumentation Supplier
3.1.3 Chemical/Reagent Supplier
3.1.4 Pathology Supplier
3.1.5 Independent Clinical Laboratory
3.1.6 Public National/regional Laboratory
3.1.7 Hospital Laboratory
3.1.8 Physicians Office Lab (POLS)
3.1.9 Audit Body
3.1.10 Certification Body
3.2 Understanding Antimicrobial Resistance
3.2.1 What is Antimicrobial Resistance (AMR)
3.2.2 Bacteria and Other Microbes
3.2.3 The History of Antibiotics
3.2.4 The Role of Animal Husbandry
3.2.5 The Implications of Horizontal Transfer
3.2.6 The Threat of AMR
3.3 The Changing Road to New Antibiotics & Technologies
3.4 The Key Role of Diagnostics in AMR
4 The Market Opportunity of AMR
4.1 The Key Large Market Opportunities in AMR
4.1.1 Streptococcus Pneumoniae (DRSP)
4.1.2 Campylobacter (DRC)
4.1.3 Clostridium Difficile (CD)
4.1.4 Staphylococcus aureus (MRSA)
4.1.5 Neisseria gonorrhoeae (DRNG)
4.1.6 Salmonella (DRNTS)
4.2 Diagnostic Technology Development Opportunities
4.2.1 What's Wrong with Microbiology
4.2.2 The Features Battleground of Infectious Disease Diagnostics
4.2.3 Multiplex vs. POC/Rapid
4.2.4 The Miracle of Genetics
4.2.5 From Multiple Pathogens to All Pathogens - The Next Next Generation
4.2.6 Gene Sequence Diagnostics WITHOUT the Sequencing.
4.2.7 Markers of Resistance.
4.2.8 What Happens to the Microbiology Lab?
5 Antibiotic Resistance Diagnostics Recent Developments
5.1 Antibiotic Resistance Recent Developments
5.1.1 Importance of These Developments
5.1.2 How to Use This Section
5.2 FDA Clears New bioMerieux Diagnostic System and Panel
5.3 Selux Diagnostics Phenotyping System Gets FDA Approval
5.4 New diagnostics to tackle drug resistant infections
5.5 Takara Bio USA, Inc. and BioExcel Diagnostics Partner for Infectious Disease Panels
5.6 Three companies join forces to tackle antimicrobial resistance (AMR)
5.7 T2 Biosystems Exercises BARDA Option
5.8 OpGen to Ramp up Use of Acuitas AMR Gene Panel
5.9 BD Partners With Pfizer, Wellcome to Study AMR Dx
5.10 Sepsis Test Developers Accelerate Plans
5.11 OpGen Receives FDA Clearance for AMR Panel
5.12 Visby Medical - Rapid Uptake of Handheld PCR Test for STIs
5.13 MicroGenDx, OrthoKey Clinic and OrthoKey Surgery
5.14 BioMerieux Receives CE Mark for Vitek Clinical Microbiology System
5.15 Hologic to Acquire Mobidiag
5.16 Campylobacter strains exchange genes
5.17 Disinfection spreads antimicrobial resistance
5.18 Molzym, Fraunhofer Developing Rapid Sepsis Dx
5.19 Illumina, IDbyDNA Build NGS-Based Respiratory Panel
5.20 Accelerate Diagnostics Expands AMR Testing
5.21 Rapid diagnostics linked to optimal antibiotics
5.22 Visby Medical Wins AMR Diagnostic Competition
5.23 DNAe Technology SARS-CoV-2 Sequences
5.24 Infections with foodborne bacteria becoming harder to treat
5.25 Dust is sharing antibiotic resistance genes
6 Key AMR Diagnostics Companies
6.1 1928 Diagnostics
6.2 Abacus Diagnostica
6.3 Abbott Laboratories
6.4 Accelerate Diagnostics
6.5 ADT Biotech
6.6 Beckman Coulter Diagnostics
6.7 Becton, Dickinson and Company
6.8 Binx Health
6.9 bioMerieux Diagnostics
6.10 Bio-Rad Laboratories, Inc.
6.11 Cepheid (Danaher)
6.12 Curetis N.V. / Curetis GmbH
6.13 Day Zero Diagnostics.
6.14 Enzo Biochem
6.15 Eurofins Scientific
6.16 Fusion Genomics.
6.17 GeneFluidics
6.18 Genetic Signatures
6.19 Great Basin Corporation
6.20 Hologic
6.21 Hutman Diagnostics
6.22 Inflammatix
6.23 Linear Diagnostics.
6.24 Lumos Diagnostics
6.25 Millipore Sigma
6.26 OpGen
6.27 Ortho Clinical Diagnostics
6.28 Perkin Elmer
6.29 Qiagen
6.30 Roche Molecular Diagnostics
6.31 SeLux Diagnostics
6.32 Sense Biodetection
6.33 Siemens Healthineers
6.34 Sysmex
6.35 Thermo Fisher Scientific Inc.
6.36 Visby Medical
7 The Global Market for Antimicrobial Resistance Diagnostics
7.1 Global Market Overview by Country
7.2 Global Market by Technology - Overview
7.3 Global Market by Technology - Overview
8 Global Antibiotic Resistance Diagnostic Markets - By Pathogen
8.1 Drug Resistant Streptococcus Pneumoniae - DRSP
8.2 Drug Resistant Campylobacter - DRC
8.3 Clostridium Difficile - CD
8.4 Methicillin Resistant Staphylococcus Aureus - MRSA
8.5 Drug Resistant Neisseria Gonorrhoeae - DRNG
8.6 Drug Resistant Salmonella - DRNTS
9 Global Antibiotic Resistance Diagnostic Markets - by Technology
9.1 Microbiology Culture
9.2 Immunoassay
9.3 PCR
9.4 NGS
9.5 Mass Spectrometry - MS
9.6 Rapid and Point of Care - Rapid/POC
10 Vision of the Future of AMR Diagnostics
11 Appendices
11.1 United States Medicare System: Clinical Laboratory Fees Schedule
For more information about this report visit https://www.researchandmarkets.com/r/fuzpmi
About ResearchAndMarkets.com
ResearchAndMarkets.com is the world's leading source for international market research reports and market data. We provide you with the latest data on international and regional markets, key industries, the top companies, new products and the latest trends.
诊断试剂
2025-01-15
SAN JOSE, Calif.--(
BUSINESS WIRE
)--Takara Bio USA Holdings, Inc. (“TBUSH”) today announced the acquisition of Curio Bioscience, a pioneering company in the field of spatial genomics. This strategic acquisition combines two innovative spatial biology platforms with Takara Bio’s industry-leading portfolio of single-cell genomics tools.
TBUSH is a wholly owned subsidiary of Takara Bio Inc. ("Takara Bio"), a leading global biotechnology and life science company headquartered in Shiga, Japan.
Takara Bio USA
, Inc. ("TBUSA") is a wholly owned subsidiary of TBUSH. TBUSA and TBUSH are part of the global Takara Bio Group, which offers diverse life science products and services supporting discovery, translational, and clinical scientists in advancing their research.
The
Curio Bioscience
acquisition will extend the power of Takara Bio’s NGS solutions and give customers deeper insights into tissue spatial organization and molecular composition. “We are proud of our history of innovation in the single-cell genomics market, having developed the first commercially available kits for single-cell RNA- and DNA-seq,” said Carol Lou, President and CEO of Takara Bio USA. “The acquisition of Curio Bioscience continues this legacy with the addition of Trekker, the first truly single-cell spatial technology.”
Curio’s advanced Trekker and Seeker technologies integrate spatial information with molecular data, enabling researchers to transform single-cell sequencing data into spatially resolved maps. This transformation offers high-resolution insights into the organization and function of cells within their native tissue environments.
“Curio Bioscience is dedicated to creating innovative approaches that map the entire transcriptome with unparalleled sensitivity and resolution,” said Stephen Fodor, co-founder and CEO. “Combining Curio’s technology with Takara Bio’s vast NGS and single-cell tool set will provide customers with industry-leading solutions for their spatial biology needs.”
Through these advancements, Takara Bio has solidified its position as a leader in the single-cell genomics market by continually driving innovation and supporting groundbreaking research in the life science market for a wide range of applications in cancer biology, neuroscience, developmental biology, and immunology.
Advisors
Houlihan Lokey acted as exclusive financial advisor and Cooley LLP acted as legal advisor to Takara Bio.
Aquilo Partners, L.P. acted as financial advisor and Dorsey & Whitney LLP as legal advisor to Curio Bioscience.
About Takara Bio
Takara Bio USA, Inc. is a wholly owned subsidiary of Takara Bio Inc. that manufactures and distributes kits, reagents, and instruments for the life sciences, including NGS, PCR, gene delivery, genome editing, stem cell research, nucleic acid and protein purification, and automated sample preparation.
Takara Bio Inc., a world leader in biotechnology research and development, offers a host of life science research solutions, from enzymes and GMP-grade reagents to contracted cell and gene therapy manufacturing services and is the developer of the RetroNectin® reagent, a world standard in gene therapy protocols. Takara Bio is committed to preventing disease and improving the quality of life for all people through the use of biotechnology. For more information, visit
takarabio.com
.
About Curio Bioscience
Curio Bioscience is advancing a new generation of high-precision tools for the life sciences industry. The company has developed innovative spatial biology capabilities to map the whole transcriptome at high resolution using existing sequencing workflows and instrumentation. The founding team has a strong track record of bringing genomic solutions to market.
Curio Bioscience
is based in Palo Alto, California.
并购
100 项与 Takara Bio USA, Inc. 相关的药物交易
登录后查看更多信息
100 项与 Takara Bio USA, Inc. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月12日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
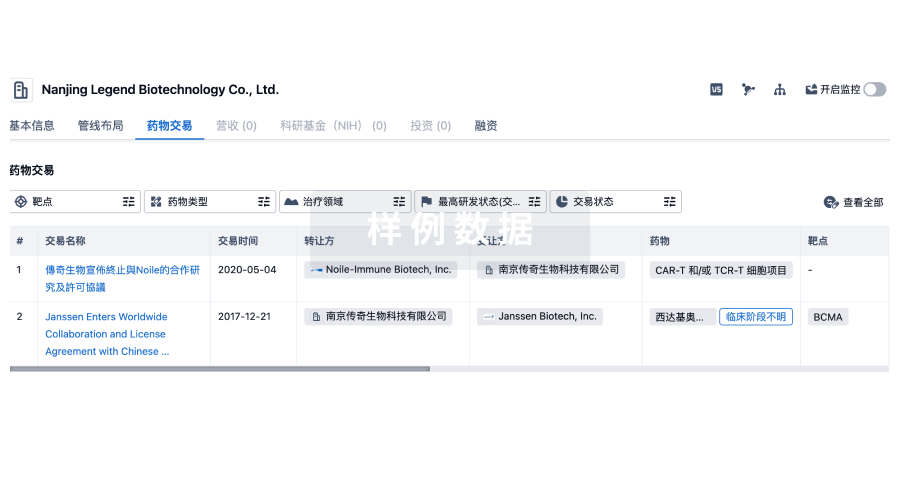
转化医学
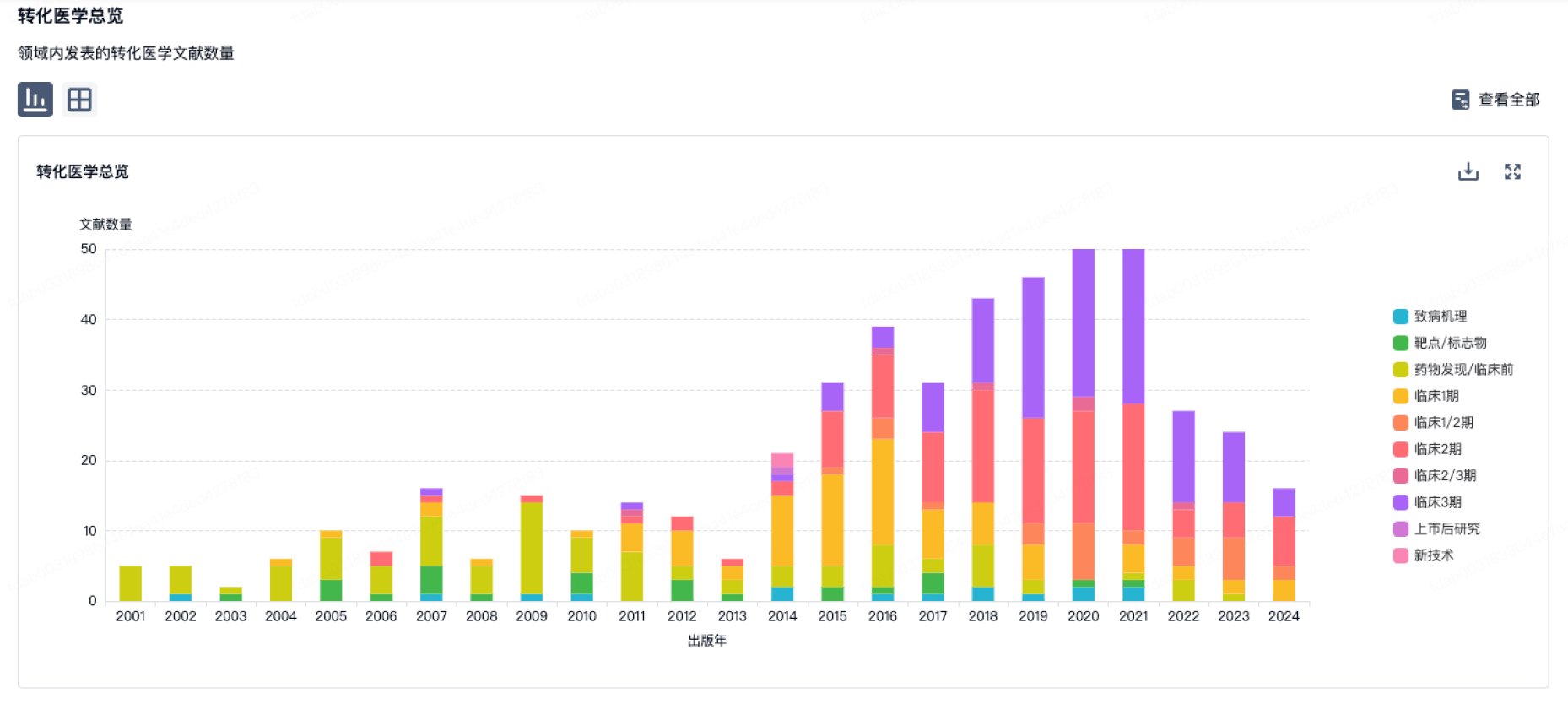
使用我们的转化医学数据加速您的研究。
登录
或
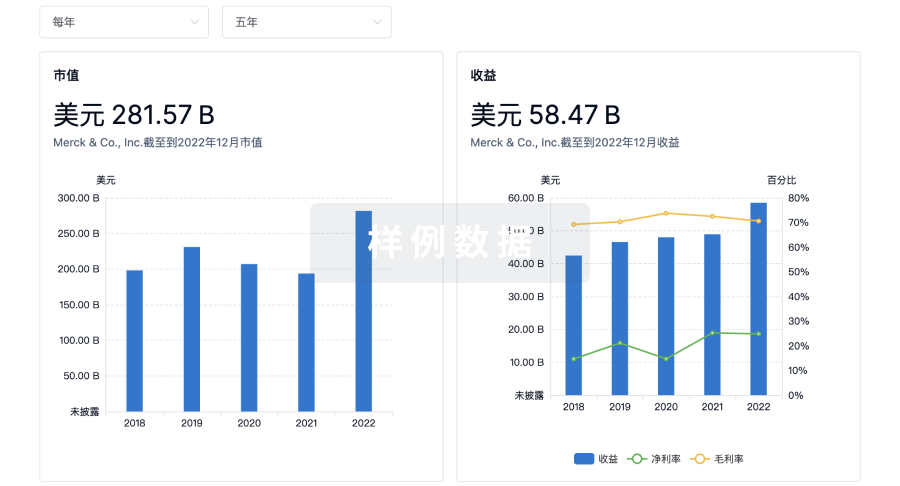
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
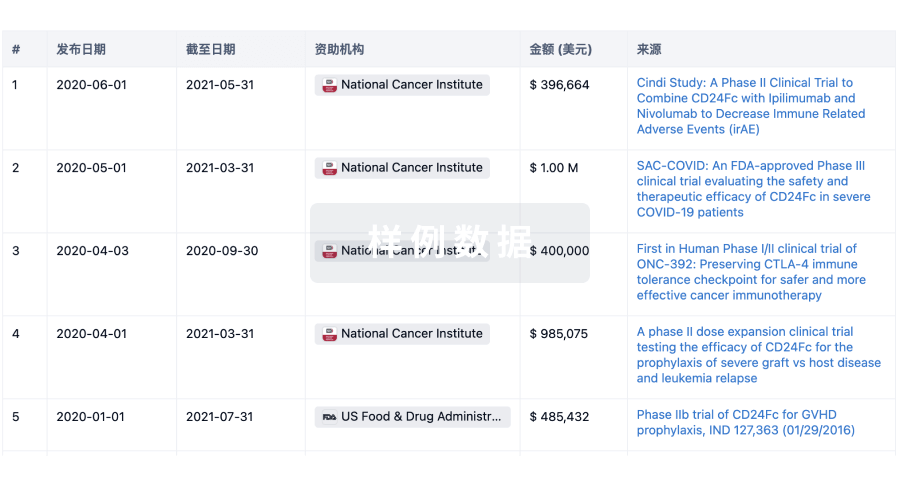
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用