预约演示
更新于:2025-05-07
Shandong Tesla Biotechnology Co., Ltd.
私营公司|2014|中国山东省
私营公司|2014|中国山东省
更新于:2025-05-07
概览
关联
100 项与 Shandong Tesla Biotechnology Co., Ltd. 相关的临床结果
登录后查看更多信息
0 项与 Shandong Tesla Biotechnology Co., Ltd. 相关的专利(医药)
登录后查看更多信息
74
项与 Shandong Tesla Biotechnology Co., Ltd. 相关的新闻(医药)2025-02-25
BFC Group简介
BFC Group是一家立足于中国的精品投行,专注于医疗健康领域的并购、专利许可、融资。我们的业务板块涉及中国、美国和欧洲,曾与国内外众多领先的医疗健康公司有过合作并保持着良好的关系。随着在美国和中国一线运营经验的不断积累,我们对客户在本土之外建立公司或扩展业务所面临的挑战有着深刻的理解。
立于塔顶,高瞻之见。
使命愿景
紧跟医疗健康领域趋势,通过合作推动科学突破与创新。
依客户需求制定战略拓展计划,并在目标市场全方位落实与执行。
成为专注医疗健康行业最优秀的投资银行。
No.1
香港资本市场
香港股市总体概览
港股持续回暖,上周恒生指数累计上涨3.8%。
2025年至今,恒生指数累计上涨17.0%。
上周,恒生医疗保健指数(HSHCI)累计上涨8.7%。
2月19日,国务院办公厅发布关于转发商务部、国家发展改革委《2025年稳外资行动方案》的通知。其中提出,推动生物医药领域有序开放。支持符合条件的外资企业参与生物制品分段生产试点,加快省级试点方案、质量监管方案审核,推动生物医药产业优化资源配置,及时协调解决试点过程中企业遇到的困难问题。研究完善医药领域开放政策,便利创新药加快上市,优化药品带量采购,进一步提高医疗器械产品采购可预期性。另同日有报道指出,从国家医保局了解到今年我国将全面推进基本医保基金即时结算改革,以加快医疗机构和医药企业资金周转。
2月19日,歌礼制药发布公告,宣布其小分子口服GLP-1R激动剂ASC30在美国的Ib期多剂量递增研究取得积极期中结果。每日一次服用ASC30口服片治疗28天后,MAD队列2 (2mg,10mg,20mg,40mg) 体重相对基线平均下降6.3%。歌礼制药-B周累计涨幅超50%。
2025年至今,医药板块领先大盘,恒生医疗保健指数累计上涨21.0%,高于恒生指数4.0个百分点。
香港股市医疗健康板块概览
2025年1月恒生医疗保健指数上涨0.4%。上周市值前20个股大多上涨。CXO概念股持续走高。
康方生物上周累计上涨14.5%,消息面上,2月14日,公司宣布其自主研发的靶向IL-4Rα/ST2双特异性抗体AK139的新药临床试验申请(IND)已获中国国家药品监督管理局(NMPA)正式受理,针对呼吸系统及皮肤疾病等多领域疾病展开探索。AK139是该公司非肿瘤领域首个进入临床阶段的双特异性抗体新药。作为IL-4Rα/ST2双抗药物,AK139可同时阻断IL-4、IL13(通过结合IL-4和IL-13受体复合物共享的IL-4Rα亚单位)和IL-33/ST2介导的炎症通路,在临床前研究中展现了优异的靶点协同效应。
香港股市医疗健康板块证券发行
2024年共计有12家企业上市(其中3家为依照18A新规上市的未盈利生物医药公司),共募集金额54.0亿港元。2025年截至目前,共有1家企业上市(其中1家为依照18A新规上市的未盈利生物医药公司),数量与去年同期持平,金额总数上领先去年同期3.6%。
上周无新增IPO。
2024年共计19家企业完成定增,共募资金额80.9亿港元。2025年至今,共有4家企业完成定增,目前数量领先去年同期33.3%,金额总数上领先去年同期975.0%。
2月21日,泰凌医药公告称已完成配售4.1亿股,配售价为每股0.33港元,筹资总额为1.35亿港元。募资用途说明:将通过抵销贷款协议A项下股东贷款的未偿还本金额及应计利息87,000,000港元偿付;将通过抵销贷款协议B项下贷款的未偿还本金额及应计利息48,351,648.35港元偿付。
香港股市医疗健康板块IPO动态
港股医疗板块拟上市企业
No.2
沪深资本市场
沪深股市总体股市概览
A股主要指数集体上扬,科创50涨近6%。AI硬件题材全线大涨,服务器、算力方向领涨,寒武纪涨停;电信股大幅反弹,中国联通、中国电信均涨停;阿里云、机器人等亦表现活跃。市场成交额时隔两个月突破2万亿元。
本周,上证指数累计涨0.97%,创业板指涨2.99%,均录得周线三连阳。
截至目前,2025年上证综指上涨0.8%,沪深300上涨1.1%。
上周,上证医药上涨3.4%,医药中信指数上涨1.9%。
2月20日晚,《支持创新药高质量发展意见》第二版征求意见稿开始在业内流传。第二版征求意见稿包括鼓励商业健康保险公司扩大创新药投资资金规模、探索新上市药品首发价格自评机制等内容,进一步厘清市场重点关注的商保丙类目录方面相关政策,短期可落地性明显增强。
2025年,A股医药板块回暖。上证医药上涨5.5%,领先上证综指4.7个百分点。医药中信指数上涨3.9,领先沪深300指数2.8个百分点。
沪深股市医疗健康板块概览
2023年医药中信指数下跌6.77%。继2024年12月下跌5.5%后,2025年1月下跌3.8%。上周市值前20个股涨跌互现。
2025 ASCO GU期间,恒瑞医药公布了Nectin-4 ADC HR-A2102治疗尿路上皮癌I期研究结果。共纳入81例尿路上皮癌患者,30%以上的患者接受过≥2线既往系统治疗,既往接受过Her2 ADC治疗比例超过35%。
SHR-A2102在6mg/kg和8mg/kg剂量组观察到较高的抗肿瘤活性。6mg/kg剂量组患者的ORR、DOR分别为41.9%、7.6个月,8mg/kg剂量组ORR、DOR分别为50.0%、5.5个月。6mg/kg和8mg/kg剂量组中位PFS均为5.8个月。在接受过Her2 ADC治疗的患者中,6mg/kg剂量组确认的ORR为54.5%,8mg/kg剂量组确认的ORR为42.9%。
沪深股市医疗健康板块概览
上周,创新药指数上涨3.6%,医疗器械指数上涨1.4%。
2月17日,华东医药宣布HDM1005注射液在中国Ia及Ib期临床试验中取得了积极结果。HDM1005注射液是是多肽类GLP-1R和GIPR的双靶点长效激动剂。
在Ia期中,体重下降呈剂量依赖性,在第8天时5mg组体重最大平均下降为3.92kg(降幅:5.36%)。在Ib期中,经过4周治疗,0.5-4.0mg组在第29天时的体重平均下降4.79-7.76kg(降幅:6.12%-10.29%),并且在停止治疗后4周体重下降仍可维持。在HDM1005组中,到第4周时体重下降>5%的受试者比例为87.5%,而安慰剂组为25%。
沪深股市医疗健康板块新股发行
2024年,共4家企业申报IPO。总体数量同比减少71.4%。共募集资金2.47亿元人民币,总体金额同比减少87.7%。
上周无新增IPO。
2024年,有7家企业发行定增,总体数量同比减少65%。累计定增金额4.95亿元人民币,总体金额同比减少67.6%。
上周新增一起定增来自主营脊柱类植入耗材和创伤类植入耗材的三友医疗。公司以13.1元/股价格发行1640万股,随后在2月19日作价4.16亿元通过发行股份及支付现金的方式收购水木天蓬37.1077%股权及上海还瞻100%出资份额。
沪深股市医疗健康板块IPO动态
沪深股市医疗板块IPO排队企业情况
No.3
NASDAQ
美国股市总体股市概览
当地时间上周五,美股大型科技股全线下跌,特斯拉跌超4%,英伟达跌逾4%,亚马逊跌超2%,谷歌跌逾2%,微软跌超1%,脸书跌逾1%,苹果跌0.11%。在特朗普出台的一系列关税相关政策后,美国消费者对长期通胀率的预期升至30年来最高水平。
美股三大指数上周全线收跌,道指周内累计下跌2.51%,标普500指数周内下跌1.66%,纳指周内下跌2.51%。
2025年截至目前,纳指累计上涨1.1%。
上周纳斯达克生物技术指数 (NBI)逆势上涨1.5%。
2月20日,阿斯利康正式宣布将以约1.6亿美元收购珐博进中国。根据协议条款,珐博进将获得8500万美元的企业价值,以及交易交割时珐博进中国持有的约7500万美元现金,总计约1.6亿美元。该交易预计将于2025年中期完成交割。交割完成后,阿斯利康将获得罗沙司他在中国的所有权利。珐博进保留其在美国及未授权给安斯泰来的市场对罗沙司他的权利。
2024年截至目前,NBI累涨6.5%,反超纳指5.4个百分点。
美国股市医疗健康板块概览
2025年开年首月,纳斯达克生物技术指数 (NBI)表现稳定,月内总体上涨5.2%。上周NBI指数强势攀升,指数成分公司股价大多上涨。
百时美施贵宝宣布了POETYK PSO长期扩展试验的五年数据,显示Sotyktu(deucravacitinib)在治疗中重度斑块型银屑病方面具有一致的安全性和持久的反应率。该试验涉及超过5000个患者年暴露,未观察到新的安全信号。临床反应率,包括PASI 75和PASI 90,从第一年到第五年保持稳定。这些发现支持Sotyktu作为银屑病潜在口服标准治疗的角色。
美国股市医疗健康板块新股发行
2024年全年NGS&NGM医疗健康板块累计有30支新股发行,累计募资超55.13亿美金。
2025年截至目前,NGS&NGM医疗健康板块共有7支新股发行,累计募资7.66亿美元,相较去年同期数量上下降22.2%,金额下降73.9%。
上周2月7日,美股NASDAQ GM板块新增两个IPO。分别是专注于为药品和医疗保健服务提高解决方案的Wellgistics Health,募资总额为450万美元。和一家总部位于德国的体外诊断产品开发商,总募资800万美元。
2024年共有322家企业完成定增,累计募集资金约293.8亿美元;2025年截至目前共有60家企业完成定增,总体数量上比去年同期下降20%,金额较去年同期下降73.2%。
上周共有9起新增定增。其中募资金额最高的是研发肿瘤免疫疗法的美国生物技术公司Arcus Biosciences。公司此次以11美元每股的价格募集了1.5亿美元。
医疗健康板块IPO动态
No.4
近期融资事件
带量采购专利到期ASCO会议
2025-02-20
·中国药闻
来源:新华网
有序扩大自主开放、提高投资促进水平、增强开放平台效能、加大服务保障力度,《2025年稳外资行动方案》2月19日对外发布,从4方面提出了20条措施。
当前,外部环境不确定性增多,我国吸引外资面临多重困难和挑战,行动方案以务实举措释放出进一步扩大开放、加大力度吸引外资的明确信号。
一系列举措在外资准入限制上做“减法”。方案提出,适时进一步扩大电信、医疗领域开放试点;研究制定有序扩大教育、文化领域自主开放实施方案,适时对外公布并稳步实施;便利创新药加快上市,优化药品带量采购;抓好《外国投资者对上市公司战略投资管理办法》落实……行动方案在外商关切领域明确了“任务书”。
在营商环境上做“加法”。方案提出,持续优化营商环境,切实把外资企业的国民待遇落实到位。研究制定鼓励外资企业境内再投资政策措施,促进外资企业在华所获利润更多用于再投资。允许外商投资性公司使用境内贷款开展股权投资,为跨国公司在华投资设立总部型机构提供便利。优化外资并购规则和并购交易程序,完善并购管理范围,降低跨境换股门槛等。
“高质量”是行动方案的关键词。一系列举措着力在利用外资上稳存量、促增量、提质量。方案明确,修订扩大鼓励外商投资产业目录,优化外商投资结构,促进外资服务我国制造业高质量发展,引导外资投向现代服务业,支持外资更多投向中西部和东北地区。
引资结构不断优化是当前我国吸引外资的重要特征。2025年1月,我国高技术制造业实际使用外资占全国实际使用外资的12.5%,较2024年全年提高0.8个百分点。医药制造业、科技成果转化服务实际使用外资分别增长68.4%和23.9%。
方案还提出,鼓励外商投资养殖、饲养设备生产、饲料兽药生产等畜牧业相关领域并享受国民待遇。支持外资企业参与我国新型工业化进程,重点引进高技术领域外商投资,为外资企业提供更多市场机会和合作空间。鼓励养老服务、文化和旅游、体育、医疗、职业教育、金融等服务业领域吸引利用外资,满足多层次服务消费需求。
在加大服务保障力度方面,行动方案立足保护外商投资权益,提升外资企业贸易便利化水平,让外资企业安心、放心、有信心。
行动方案明确,支持将更多外资项目纳入重大外资项目和重点外资项目清单;尽快制定出台相关文件,明确政府采购本国产品标准,确保不同所有制企业在中国境内生产的产品平等参与政府采购活动;加快互免签证协定商谈,继续稳妥扩大单方面免签国家范围。
2025年2月11日,特斯拉上海储能超级工厂在上海自贸区临港新片区正式投产。新华社记者 方喆 摄
新年伊始,开放的春风温暖人心,一批外商投资项目“新”意盎然。
特斯拉上海储能超级工厂正式投产、跨国制药企业赛诺菲的生物原料药项目正在北京加快推进中、西门子医疗全新研发制造基地项目在深圳奠基……开年以来多个外资大项目落地,高端化、智能化、绿色化特点鲜明,彰显外资看好中国发展前景的坚定信心。
商务部当天发布数据显示,2025年1月,我国实际使用外资金额975.9亿元,环比增长27.5%。同期,英国、韩国、荷兰、日本实际对华投资分别增长324.4%、104.3%、76.1%、40.7%。
随着2025年各项举措落地见效,一个致力于实现高质量发展、坚定开放合作的中国,必将为世界注入更多信心和动力、带来更多合作共赢新机遇。
带量采购核酸药物并购
2025-02-17
新旧动能转换,体现在标志人物命运变迁上。当王石开始探店带货养家时,已经无人在意过去两年梁文锋还扛着量化大镰刀。
马云俨然是行走的信号发射塔,重新露面,叠加着中国科技资产重估的宏大叙事。
瑞银给出一份中美科技股对标名单:寒武纪对标英伟达,小米对标特斯拉,中芯国际对标台积电,腾讯对标Meta,阿里巴巴则对标亚马逊,百度对标谷歌,中兴通讯对标思科。
瑞银的本意应该是引导西方投资者,更准确理解中国科技公司的业务模式和潜力。
华泰证券给出一份中国科技股七巨头名单:以苹果、谷歌、亚马逊、微软、Meta、特斯拉、英伟达为代表的科技七巨头(Magnificent 7),已成为美股科技核心资产,展望未来,小米、联想、比亚迪、中芯国际、阿里巴巴、腾讯、美团有望成为中国科技核心资产。
争议最大的是联想(AI端侧落地核心公司)。几个菜啊?喝成这样。
科技重估的风正吹到医药。虽然生物科技产业在国内地位不高,但墙内开花墙外香,自2015年药审改革发端,仅用10年,中国已成为世界医药创新的源头之一。
中国创新药企崛起的速度远超当年日本,他们有武田制药、第一三共,我们也将有自己的MNC。
还有更大的奇迹还在后面,基于两个逻辑。一是创新药迄今的成就,是在激励不足的情况下达成的,意味着后劲很大,一旦外部环境边际改善,将升势汹涌;二是创新药仍在发展早期,终局未定,仅有两三家药企成为巨头的预期比较明朗,一大批Biotech都有勇立潮头的机会。
时间周期:2023年4月-2024年3月 数据来源:AnswersNews
01
最被低估的中国科技资产
迈瑞医疗逼近全球医疗器械前20名,药明系冲击全球CXO前三,都在掀开国际化的史诗级征途。限于篇幅,今天只聚焦创新药巨头的诞生。
在老龄化及控费背景上,日本药企与我们相似。上世纪80年代最高峰时,日本曾有超过1700家医药企业,到2015年仅剩下305家,超过五分之四的药企被出清。踩着同行的尸骨,少数巨头崛起。据美国《Pharm Exec》杂志数据,2023财年武田制药处方药销售额/研发投入为276.87亿美元/47.76亿美元,而恒瑞医药处方药销售额/研发投入为32.32亿美元/10.93亿美元,完全不是一个量级的,武田的研发投入甚至远超恒瑞医药的销售额。
日本巨头似乎遥不可及,但我们变换一下视角,中国生物科技在创新活力及Biotech生态上其实已经迅速实现反超。
据医药魔方,2016年中国创新药核心临床试验开展数量为103项,处于中美英日第4位,且大幅落后于美国,相比英日亦有较大差距,但至2022年中国已增长至322项,与美国的339项几乎并驾齐驱,并大幅领先于英国和日本。
到2024年,中国ADC、细胞疗法、双抗、溶瘤病毒等新兴疗法管线数量占比均跃居全球第一。
资料来源:医药魔方,华源证券研究所
这并不是简单粗暴的堆量,其管线创新含量得到MNC用脚投票的认可。
据DealForma数据,2024年大型跨国药企引进的约31%创新药候选分子来自中国,2019年这一数字为0%。据医药魔方,2024年国产创新药对外BD在全球BD中的总金额占比达30%,2019年这一数字还是0%。
海内外不同数据来源都指向同一事实。
相比之下,日本创新生态却暮气沉沉,Biotech产业严重滞后。据海通国际,在2017-2022年FDA获批药物中,美国约一半获批药物由1990年后成立的公司创造,而日本所有获批药物均为1980年前的老牌药企创造。
日本错失生物药浪潮。在美国TOP10畅销药物榜单中,2000年和2010年上榜药物全部为化学药,日本药企均有上榜产品,以大冢制药研发的阿立哌唑为代表,其美国销售额在2010年达到45亿美元。2020年后,绝大多数上榜药品为生物药,而日本在生物药领域声势渐微。背后原因错综复杂,其中核心原因之一是长期医保控费下本土市场逐渐失去创新活力。
中国创新药企的生存韧性却异常强大。
据青侨阳光,以下图统计的公司为例,用2023年的研发开支除以在研管线数量,可得出每10亿美元支撑的研发管线数量,欧美头部药企普遍是15-16款左右,日本药企典型是30+款,而中国药企平均有100+款,超过欧美MNC的6倍!
低成本高效率,天下无敌。
于是,在投融资及控费寒冬中找到自适应生存之道,BD/M&A成为创新药现金流主要来源。据医药魔方、华泰研究,截至11 月18日,2024年国内创新药企业直接融资金额约为39.77亿美元(剔除IPO及IPO后融资后,约为25.63亿美元),而创新药BD和M&A的首付款/收购款(不含里程碑收入)高达62.73亿美元,反超直接融资成为主要资金增量,且潜在总金额达432.04亿美元。
木头姐强调,医疗保健是最被低估的AI应用,今天我们通过横向、纵向对比,也想提醒生物医药是最被低估的中国科技资产,尽管过去3年处境艰难,但其快速进步及全球地位已远超公众的认知。
02
我命由我不由天
创新药是被压到极致的弹簧,细微改善也将释放可观弹性。
医保商保一体化同步结算平台已经开始上线运行,目前已实现山东省全省上线,在推进基本医保与定制型商业医疗保险的协同对接上出现实质性进展。
春节后医保局表示将进一步完善集采政策, “对于预计投标企业数量超过一定规模、竞争比较激烈的品种,提前进行强竞争预警,提示企业慎重决策,科学投标,理性报价。”
预计公众将在今年上半年第11批国采及提前启动的医保谈判中,看到“理性报价”的情况。
激励创新愈发成为一种全局的正确性,AI医疗点燃医药春天的第一把火,我们还可以看得更远,3家创新药企具备成为巨头的潜质。
作为一家成立28年的传统药企,恒瑞医药并没有困于路径依赖,始终努力跟上时代的节拍。
恒瑞推开武田这扇窗,看见的将是一片海。2023财年,恒瑞医药营收与武田本土市场收入相当,而武田本土市场收入占比仅有约10%。
恒瑞应该在海外再造数个自己,其立足内需的发展模式接近极限,即使每款创新药都是国内前三,但在每个细分方向上面临着中小药企的贴身肉搏,不存在断层式领先。新上市的增量品种需要抵消老化品种销售额的下滑,有可能出现营收原地踏步的窘况。
所以,即使港股IPO拉低估值,恒瑞也必须国际化蝶变重生。
作为自主出海的代表,百济神州将坐稳创新药业务收入第一的位置,泽布替尼有望跻身30亿美元分子。
百济神州在JPM大会继续释放强劲的发展信号,CDK4i、pan-KRAS、B7-H4
ADC、EGFR PROTAC、PRMT5/MAT2A、IRAK4 PROTAC,有望成为未来的重磅产品,销售预期合计190亿美元,足以撑起MNC的体量。
信达生物全面均衡,基本无可挑剔,没有明显的缺点和风险点。
2024年产品收入超82亿元,同比增长40%以上,2024Q4产品收入22亿元,过去3个季度产品收入分别为23 亿元、20 亿元、17 亿元,总体保持平稳增长。2025年是商业化大年,主要解锁高潜慢病市场空间,成为公司第二增长极。PCSK9单抗纳入新版国家医保目录并已于2025年1月1日生效,玛仕度肽(GCG/GLP-1双受体激动剂)、替妥尤单抗(抗IGF-1R单抗)及匹康奇拜单抗(抗IL-23p19单抗)三大慢病单品,也将在年内上市。
2025年也是信达的国际化BD大年,预计不定期催化剂、灵活多变的出海形式将贯穿全年。
仅仅10年,中国创新药即收获了一大片森林,有理由相信未来会诞生多家国际化大型药企。
创新药的发展进程是非线性的,一次大额BD或大型并购,都有可能改变一家药企的命运,所谓我命由我不由天,期待从后排杀出黑马。
创新药永远有奇迹。
医药出海
100 项与 Shandong Tesla Biotechnology Co., Ltd. 相关的药物交易
登录后查看更多信息
100 项与 Shandong Tesla Biotechnology Co., Ltd. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月17日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
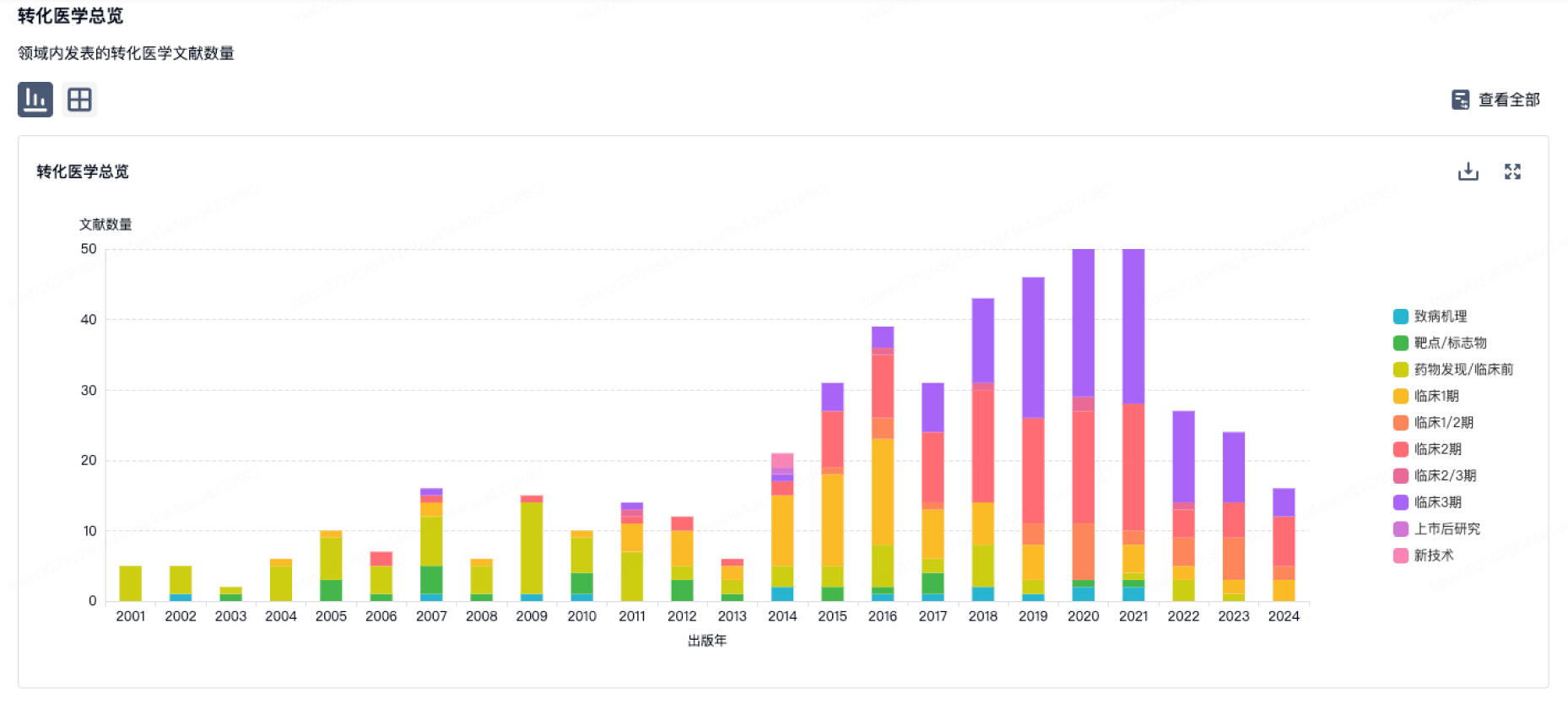
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用