预约演示
更新于:2025-05-07

Nuevolution AB
更新于:2025-05-07
概览
关联
5
项与 Nuevolution AB 相关的药物作用机制 凝血因子抑制剂 |
在研机构- |
原研机构 |
在研适应症- |
非在研适应症 |
最高研发阶段无进展 |
首次获批国家/地区- |
首次获批日期- |
靶点 |
作用机制 BET抑制剂 |
在研机构- |
原研机构 |
在研适应症- |
最高研发阶段无进展 |
首次获批国家/地区- |
首次获批日期- |
靶点 |
作用机制 αvβ3拮抗剂 |
在研机构- |
原研机构 |
在研适应症- |
非在研适应症 |
最高研发阶段无进展 |
首次获批国家/地区- |
首次获批日期- |
100 项与 Nuevolution AB 相关的临床结果
登录后查看更多信息
0 项与 Nuevolution AB 相关的专利(医药)
登录后查看更多信息
5
项与 Nuevolution AB 相关的新闻(医药)2025-03-31
·先导药物
国泰君安发布研报称,AI 技术对 CXO 产业范式革新的渗透正在加速,其在生物医药领域的深度应用有望重塑 CXO 产业价值链。美股医疗 AI 代表公司 Tempus AI 单周涨幅约 30%,年内涨幅超 160%,持续推动医疗 AI 概念升温。在此大背景下,成都先导作为 AI 医疗领域的重要参与者,其动态备受瞩目。近期其业绩飘红引发了广泛关注,这一现象背后,折射出CRO行业的发展趋势。图源:成都先导官网业绩大幅增长,AI赋能成效初显Guide View成都先导于 2 月 27 日晚披露 2024 年度业绩快报,报告期内实现营业收入 42,698.69 万元,同比增长 14.99%;归属于母公司所有者的净利润 5,093.66 万元,同比增长 25.09%;归属于母公司所有者的扣除非经常性损益的净利润 5,536.98 万元,同比增长 1,504.05%。公司财务状况良好,资产负债率低,货币资金充裕。图源:成都先导官网公司构建DNA编码化合物库技术(DEL)、基于分子片段和三维结构信息的药物设计技术(FBDD/SBDD)、寡聚核酸新药研发相关技术(OBT)和靶向蛋白降解相关技术(TPD)四大核心技术平台,形成了从靶点发现到临床申报的一体化服务能力。公司的核心技术平台和关键新药研发能力的协同效应持续放大,实现营利双增。据了解,其业务重心适度向商业项目倾斜,对自研管线项目聚焦调整,报告期内自研管线HG146正式进入临床二期。公司在报告期内的技术平台自研项目侧重在全流程整合式DEL+AI+自动化的“设计-合成-测试-分析”(DMTA)分子优化能力平台建设。图源:成都先导2024年半年度报告成都先导在AI医疗领域的技术积累与市场布局被认为具有较大价值潜力,其成长性备受关注。AI赋能新药研发:DEL技术的深度融合Guide ViewAI 驱动 CXO 产业智能化转型国泰君安指出,全球药企降本增效需求迫切,叠加 AI 技术日益成熟,促使 CXO 企业加快智能化转型步伐。像药明康德、康龙化成、凯莱英、泰格医药等头部 CXO 企业,已通过自主研发或合作方式,构建起 AI 技术壁垒。在新药研发的靶点发现、患者招募、工艺优化等关键环节,AI 的应用显著提升了效率,有力推动行业从传统 “人力密集型” 向 “技术驱动型” 转变。以药明康德为例,其基于DEL库(DNA编码化合物库)与AI毒性预测模型联动,成功实现“分子设计-体内毒理验证”闭环,临床前开发周期从18个月缩短至12个月。泰格医药利用 AI 患者动态匹配系统,整合全国 800 + 医院脱敏数据,借迁移学习技术将患者匹配精度提升至 89%,患者入组周期缩短 28%。同时,去中心化临床试验(DCT)与 AI 结合渐成趋势,具备医疗数据处理及合规能力的企业,有望借此构筑竞争护城河。不久前,康龙化成宣布,其子公司康龙化成临床研究服务有限公司已完成对海心智惠的控股交易,其为康龙集团从“全流程一体化国际化CRO,CDMO服务提供商”向“数据和AI赋能的CRO,CDMO服务商”升级的重要一环。在 CDMO 领域,AI 赋能可实现工艺参数实时调优与质量预测,打破传统经验驱动模式,如凯莱英经强化学习优化关键参数,使某抗病毒药物中间体合成收率从 72% 提至 88%,批次稳定性从 8.5% 降至 2.3%。这些实例充分表明,AI 技术已成为 CXO 企业增强竞争力的核心要素,引领行业迈向全新发展阶段。在 CRO 行业中,顺应智能化转型趋势,积极运用 AI 技术提升服务效率与质量,是企业在激烈市场竞争中制胜的关键路径。成都先导独特的 DEL 技术成都先导成立于 2012 年,主要业务为创新药早期研发——苗头化合物和先导化合物发现与优化。公司的核心技术是DNA编码化合物库(DEL)技术,该技术结合组合化学与DNA技术,通过均分与合并增加化合物数量,构建起超大规模化合物库。2024 年上半年,DEL 板块作为公司的基石业务,保持稳中有进,实现收入 7,275.74 万元,同比增长 1.91%;2023年DEL 板块实现收入1.84亿元,同2022年比增加25.45%。作为 DEL 技术全球领先者,成都先导的 DEL 库规模和质量保持国际领先,分子数量已突破 1.2 万亿,公司持续筛选并输出高新颖性的先导化合物,筛选综合成功率近 80%。AI赋能药物研发成都先导积极探索 AI 技术与 DEL 技术的融合路径,利用 AI 优化化合物库的设计和筛选流程,以此提升新药研发效率。公司与 Cambridge Molecular(剑桥小分子)合作,在成都先导的DEL技术平台上,引入Cambridge Molecular为DEL高度优化的深度机器学习系统——DeepDELve 2,持续提高筛选效率。DeepDELve 2是一个强大且高度专业化的深度学习软件工具集,可为药物靶点高效提供具有潜在活性、更易得的化合物,其已在多个外部项目上进行过密集的使用和迭代,是 DEL + AI 分子发现领域较为先进的系统。此前,成都先导公布同腾讯AI Lab合作共同设计开发了一款分子骨架跃迁算法(GraphGMVAE),该算法可以在保持分子侧链不变的情况下,生成具有相似活性不同骨架的分子。此外,还提出了一套对分子进行优先级排序的流程,可以缩小验证范围,该结果发表于ACS Omega上。以JAK1抑制剂Upadacitinib为例,证明了该模型在药物发现中的适用性,可以像人类专家一样有效和准确地寻找新的候选分子。成都先导拥有由计算科学团队和顶级互联网公司共同设计开发的人工智能骨架跃迁平台,配合后续自建的虚拟筛选、3D-CNN对接重打分和ADMET预测平台,可以实现快速的分子评估、排序并得到候选化合物。图源:成都先导官网成都先导聚焦于AI技术与其核心技术DEL/FBDD/SBDD有效结合,对创新药小分子的发现与优化具有十分重要的意义。其计算科学 CADD/AI 团队拥有多领域人才,在 FBDD/SBDD、小核酸、靶向蛋白降解等领域,借助 AI 技术提升研发效率,如在靶向蛋白降解领域利用 CADD 加速三元复合物的优化过程。这些技术创新和融合应用,使其成为 AI 制药概念的热门标的,吸引市场关注与资金投入。在 AI 制药浪潮中的战略布局与竞争态势Guide View内部发展:多维度筑牢竞争根基成都先导积极搭建外部合作网络。2024 年 10 月,阿斯利康、百时美施贵宝、强生、默沙东、辉瑞、罗氏等国际药企携手成都先导组建全球首个DNA编码化合物库(DEL)联盟。该联盟旨在通过创新合作模式,共享资源,高效构建 DEL 库,分享最佳实践,加速药物发现与开发。成都先导凭借自身技术专长与丰富经验,作为唯一服务供应商加入联盟,为联盟的 DEL 建库项目提供支持。其亦积极推进AI技术在药物发现与优化方面的深度应用与业务布局。图源:Insight、开源证券研究所在研发投入方面,成都先导在四个核心技术平台及新药管线上持续投入。2024 年上半年研发投入 3,073.40 万元,占营业收入比例 15.78%;2024 年前三季度研发投入 5,165.20 万元,占营业收入比例 17.32%。公司拥有实力雄厚的研发团队,截至 2024 年上半年,研发人员达 392 人,是 DEL 细分领域团队规模最大的研发服务公司之一。高研发投入、壮大的团队为技术创新提供支撑。可以预见,对于成都先导等 CRO 企业和一些大型的制药公司而言,在当前市场环境下并不会选择放弃AI技术,甚至会在预算范围内进一步增加投入。外部竞争:国内国际竞争势态在国内,众多 CRO 企业如药明康德、美迪西、药石科技、成都先导、皓元医药、泓博医药等纷纷布局 AI 制药领域。其中,药明康德的 AI 算法侧重于传统 QSAR,其基于 DEL 库与 AI 毒性预测模型联动,成功缩短了临床前开发周期;泓博医药在 DEL+AI 领域布局较晚,数据积累相对不足;维亚生物则以 CRO 业务为主。在国际市场上,国际 TOP20 的药企几乎都布局了 DEL 技术,一批专注于该项技术的 CXO 企业陆续出现,如Nuevolution(2001年)、X-Chem(2009年)、成都先导(2012年)等。其中,X-Chem 是成都先导在 DEL 技术领域的直接竞争对手,在 DEL 库的构建和筛选方面经验丰富,与全球多家知名企业合作,具有一定市场影响力,但其 AI 应用商业化程度较低。图源:X-Chem官网AI能否成为CRO行业的变革引擎?Guide View政策支持与市场前景广阔政策层面,“十四五” 规划明确 AI 医疗为战略方向,2024 年医疗 AI 三类证审批周期缩短 33%,医保目录新增 12 项 AI 诊疗服务。光大证券指出,2023 年中国 AI + 医疗市场规模达 315 亿元,预计 2025 年将突破 800 亿元,复合增长率达 58.3%;全球市场更将迎来爆发,到 2030 年规模预计突破 1.5 万亿美元,其中药物研发、影像诊断、健康管理三大板块占比超 60%。未来展望与战略规划图源:成都先导官网成都先导在“ AI + 药物发现” 发现领域具备显著先发优势,随着其 “DEL+AI” 一体化平台的持续建设与完善,有望加速推进多个AI驱动发现与优化的分子进入临床验证阶段。建议重点关注其管线推进速度、MNC合作项目进展及DEL库规模扩张与迭代情况。参考来源:[1]公司官网/网络新闻[2]国泰君安[3]开源证券[4]金融界[5]投资界[6]药融圈制作策划策划:Whale / 审核校对:Jeff撰写编辑:Whale / 封面图来源:网络媒体合作 | 微信号:GuideView2021投稿转载 |13291812132(同微信)免责声明:本文仅代表作者个人观点,参考文献如上,如对文中内容有不同意见,欢迎下方留言讨论。如需转载,请邮箱guideview@guidechem.com联系我们。有问题就上「盖德问答」,点击「阅读原文」即可跳转~
财报核酸药物
2024-12-27
·医药速览
聚焦
共价
早研早聊
学以致用 侃侃而言
DOI: 10.1038/s41589-024-01791-2
前日,瑞士苏黎世联邦理工学院Andress Gloger和Jörg Scheuermann在Nature Chemical Biolgoy上发表了重要观点,回顾了DNA编码化学文库(DECL)筛选技术30多年的发展历程,在这个过程中出现了一些关键性节点、人物、和科学研究团体,并特别强调了GSK一篇开创性文章在DEL发展中的重要作用(深感认同)。
那么,今天就简明扼要地讲一个故事。
(欢迎1.25倍速“听一听”,听我亲口讲给你听)
末尾有投票,欢迎投下你喜爱的一个方向,早研早聊将根据投票结果后续着重更新。
缘起1992
1992年,Scripps研究所的Sydney Brenner和Richard A. Lerner两位教授提出一个概念性设想:以组合方式,在磁珠上合成多肽,同时通过不断增长的DNA“条形码”在同一颗粒上“记录”合成过程。
(Proc. Natl Acad. Sci. USA, 1992, 89, 5381–5383)
这个想法很快于1993年由Affymax研究所的Mark A. Gallop团队作为原理验证而付诸实践了。然而,当时存在着技术壁垒,如很难在同一固体载体上结合肽和DNA合成,因此,这种方法最终被放弃了。
(Proc. Natl Acad. Sci. USA,1993, 90, 10700)
复苏2004,“百家争鸣”
在此后,这个领域几乎沉寂了十多年,直到2004年,来自哈佛大学的David R. Liu(刘如谦)提出了新的可用想法,即,DNA模板合成法。
(Science, 2004, 305, 1601–1605)
同一时期,来自斯坦福大学的David R. Halpin和Pehr B. Harbury提出并使用了基于编码的路径法。
(PLoS Biol., 2004, 2, 1022–2030)
来自瑞士苏黎世联邦理工学院的Dario Neri提出了文库的编码自组装法。
(Nat. Biotechnol., 2004, 22, 568–574)
几年之后,学术界和工业界团队开发并公布了“拆分-合并”的DEL构建模式,也是现如今应用最为广泛的DNA编码化学文库法,没有之一。
GSK开创性研究工作,2009
其中来自GSK(早期为Praecis团队,后被GSK收购)的科学家团队,在Nature Chemical Biology上发表了第一个大型文库的设计、合成、以及应用于靶标筛选的开创性工作。这篇文章包罗万象,是DEL领域或者初学者最不容错过的“不二选”文章,其中的设计方案(Headpiece设计、DNA tags设计、小分子库设计、筛选方案设计、分析方案设计等)至今仍是最常用的方法。
这里还不得不提另一项技术的发展对DEL技术的应用的推动作用,它就是二代DNA测序(NGS)技术。这项技术的发展和普及是数以百万的深度采样实现了前所未有的低成本。NGS在DEL筛选技术中的应用也最终将该项技术推向了一个新的层面,使大型(数以亿计、兆计)DEL文库的合成和筛选成为可能以及变得更加有意义。
GSK开创性的研究成果正是在这种背景下发表的!
这篇开创性的文章中讲述了两个大型均三嗪集中型DELs的合成,包含有8亿独特的DEL分子,这比传统高通量筛选可以筛选的任何化合物集合都要大几个数量级。
如下图,他们发明了一个“发卡式”的双链Headpiece,通过酶连接进行DNA片段的高效扩展,目前已经成为DNA编码文库技术的事实上的行业标准!Headpieces通过PEG链给出游离的氨基功能片段,是小分子构建砌块组合起来的起点。
GSK利用合成的文库针对两个药理学上有趣的靶标,Aurora A激酶、和p38 MAP激酶,实现了实际上的筛选。这一刻,也标志着DEL技术在工业中用于药物发现的开始。
GSK团队的工作清楚地表明,基于DEL的亲和性筛选,在识别重要的构效关系方面具有内在的优势,有助于指导药物化学优化先导分子。
这篇文章中建立起来的DNA兼容性反应被广泛的应用,并被作为新型反应开发的样板。这篇文章暗示的该项技术的一些局限性,很大程度上,也适用于今天。
由于不同合成周期的化学反应不可能完全的转化,在组合DEL合成过程中会不断地积累截短或失败的结构。而这些结构与期望的最终全长分子编码相同。这种在化学型与编码之间的差异,最终导致基于亲和力筛选的信噪比较差,并阻碍了对真正Hits的识别。
因此,即使构建数以十亿到万亿的超大型库,理论上可行,但DEL技术目前仅限于数百万到数十亿高合成质量的成员的库,而这些库源自于不超过四个“拆分-合并”循环。
广泛认同、飞速发展
正是GSK科研团队等的工作,最终使制药行业广泛接受DEL技术用于小分子药物发现,并在内部创建团队或吸纳收购小型DEL团队,或寻求外部CRO合作。
GSK收购了Praecis (2006)
Amgen收购了Nuevolution (2019)
GHO Capital收购了X-Chem(2020)
Eli Lilly收购了Dice Therapeutics(2023)
Pfizer、Roche、BMS、Johnson&Johnson、AstraZenca、MSD成立了“DEL大联盟”(2024)
多个CRO组织,如X-Chem(2009)、HitGen(2012)、WuXi AppTec(2017)更是为制药企业和与药物研发组提供了更加贴心的FTE、FFS、以及多样性Kits服务。
就技术本身而言,由于被广泛地认可,以及可适用领域的不断扩大,也促使着DEL各项技术迅速发展。
就化学领域而言,如固体载体地可逆吸附;
(J. Am. Chem. Soc., 2019, 141, 9998–10006)
就筛选技术而言,如光亲和标记技术、微流控表型OBOC筛选、基于细胞的筛选;
(Nat. Chem.,2022, 14, 129–140.; ACS Comb. Sci., 2017, 19, 181–192.; ACS Comb. Sci., 2015, 17, 722)
就DEL构建而言,如静态和动态双药效展示DEL、集中型DEL;等。
(Trends Pharmacol. Sci., 2023, 44, 817–831)
在其它方面,一些值得注意的如,非营利性倡议正在使DEL筛选更加公开、可用,进一步促进技术的普及性。
DEL目前已经应用于小分子药物发现的所有领域,如新型的蛋白水解靶向嵌合体(PROTAC)配体,和靶向蛋白质降解的分子胶的发现,以及靶向小分子药物偶联五或放射配体的开发等。
目前已有多项来自DEL筛选的Hit发现进入了后期临床开发阶段(见如下往期回顾),更多来自DEL技术的临床候选药物有望成为可能,最终使患者收益。
往期相关精彩回顾
来自DEL的临床候选药物
2024-03-18
来自DEL的环肽苗头化合物
2024-03-21
它来自DEL——从DEL中发现的共价配体化合物
2024-04-12
无预定靶点的DEL无偏筛选
2024-03-11
G蛋白偶联受体(GPCRs)配体发现的“理想国”:基于细胞的DEL筛选方法汇总之GSK经验
2024-09-12
G蛋白偶联受体(GPCRs)配体发现的“理想国”:基于细胞的DEL筛选方法汇总之普渡大学Krusemark教授团队经验
2024-09-20
国际DEL研讨会前瞻:早研早聊分享DEL合集、及历届DEL研讨会大盘点
2024-10-14
关于PROTACs、分子胶等靶向降解剂(TPD)的高通量筛选
2024-05-24
往期链接
“小小疫苗”养成记 | 医药公司管线盘点
人人学懂免疫学 | 人人学懂免疫学(语音版)
综述文章解读 | 文献略读 | 医学科普 | 医药前沿笔记
PROTAC技术 | 抗体药物 | 抗体药物偶联-ADC
核酸疫苗 | CAR技术 | 化学生物学
温馨提示
医药速览公众号目前已经有近12个交流群(好学,有趣且奔波于医药圈人才聚集于此)。进群加作者微信(yiyaoxueshu666)或者扫描公众号二维码添加作者,备注“姓名/昵称-企业/高校-具体研究领域/专业”,此群仅为科研交流群,非诚勿扰。
简单操作即可星标⭐️医药速览,第一时间收到我们的推送
①点击标题下方“医药速览”
②至右上角“...” ③点击“设为星标
并购
2024-03-05
点击上方的 行舟Drug ▲ 添加关注小分子药物筛选技术研究现状及其应用进展来源《医药导报》 2024年2月 第43卷第2期作者武瑞君,李玮琦,杨阳,王晶,张鑫,方子寒,张小奕,苏月中国生物技术发展中心摘要小分子药物筛选技术伴随着药物的发现正在不断更新和拓展,药物筛选技术的创新可以提高研发效率和成功率、缩短研发周期、降低成本。从基于已知活性化合物和高通量筛选等传统筛选技术,到基于结构的药物发现、基于片段的药物发现、DNA编码化合物库、蛋白降解靶向联合体等新技术,小分子药物筛选技术在不断拓宽小分子药物的市场潜力。该文将介绍目前小分子药物筛选技术整体现状,系统综述各技术及其优劣势,为小分子药物筛选新技术的发展提供重要参考。关键词小分子药物筛选技术; 高通量筛选; 基于结构的药物发现; 基于片段的药物发现; DNA编码化合物库; 蛋白降解靶向联合体_正文_近年来,以抗体药物为代表的生物大分子药物研究热度持续增高,但小分子药物因具有分子量小、给药途径多、免疫原性低、可以穿透细胞膜、研发成本低、生产工艺成熟、易于储存和运输等优势[1],依然是创新药物研发的主战场。先导化合物的发现与优化在小分子药物研发过程中处于至关重要的环节,高质量的活性先导化合物能够大大缩短药物研发周期、节约成本、提高研发成功率[2]。随着生命组学、系统生物学、结构生物学等新兴学科以及高性能计算、大数据分析、人工智能等信息技术深度融入药物研发,小分子药物筛选新技术伴随着药物的发现正在不断更新和拓展,在基于已知活性化合物(Known)的药物发现和高通量筛选(high-throughput screening,HTS)[2]等传统筛选技术的基础上,基于结构的药物发现(structure-based drug discovery,SBDD)、基于片段的药物发现(fragment-based drug discovery,FBDD)、DNA编码化合物库(DNA encoded compound library,DEL)、蛋白降解靶向联合体(proteolysis targeting chimeras,PROTAC)等药物筛选新技术应运而生[3-7]。笔者在本文将介绍目前小分子药物筛选技术整体现状,系统综述HTS、SBDD、FBDD、DEL、PROTAC等技术平台及其优劣势,分析小分子药物筛选新技术研发的重要性,为小分子药物筛选新技术的未来发展提供参考。1小分子药物筛选技术整体发展现状新药研发具有周期长、投入大、风险高等特点。以小分子药物为例,研发周期平均需要约10年,包括发现苗头化合物(Hit)并经过层层结构优化得到先导化合物的药物发现阶段(2~4年)、针对候选化合物(candidate)的临床前研究阶段(1~3年)和临床阶段(3~7年)。其中,药物发现阶段是小分子药物研发中最重要的基础环节,且药物筛选技术直接关系到先导化合物质量、研发效率、研发成本以及成药可能性,是新药研发持续进行的关键。1985年之前,先导化合物的发现主要是通过人工进行,每周处理的样本数仅有数百个[8]。随着分子生物学、结构生物学等现代科学的快速发展,小分子药物发现进入基于靶点的药物设计时代,传统药物筛选方法如基于Known的药物发现由于效率低、较难研发出原创性成果、容易陷入专利陷阱等缺点,无法满足新药研发需求,药物筛选新技术不断更新迭代,HTS大大缩短先导化合物开发在药物研发中的时间,SBDD、FBDD等也逐渐成为小分子药物研发的常见手段[2-5]。通过对2022年至今在Journal of Medicinal Chemistry期刊上发表的文章进行分析统计,结果发现基于HTS、SBDD、FBDD、DEL和PROTAC的应用占比分别约为17.4%、51.2%、19.5%、4%、6.3%,在一定程度上反映目前SBDD、FBDD等新策略在新药研发领域发挥越来越重要的作用。文章内容由凡默谷小编查阅文献选取,排版与编辑为原创。如转载,请尊重劳动成果,注明【来源:凡默谷公众号】。2不同小分子药物筛选技术的最新研究进展与应用2.1 HTSHTS技术出现于20世纪80年代末和90年代初,是指以分子水平和细胞水平的实验方法为基础,以微板形式作为实验工具载体,以自动化操作系统执行试验过程,以灵敏快速的检测仪器采集实验结果数据,运用计算机对实验数据进行分析处理,并以相应的数据库支持运转的技术体系。该技术可在短时间内对数以千万的样品进行检测,具有高度标准化、筛选速度快、灵敏度高、自动化程度高、特异性强、所需样品量小等优点[9]。经过几十年的发展,HTS已经发展成为目前主流的小分子药物筛选方法之一,大量获批药物均由该技术筛选得到,例如治疗糖尿病的二肽基肽酶4(dipeptidyl peptidase-4,DPP-4)抑制剂西格列汀(sigliptin)、治疗乳腺癌的酪氨酸激酶抑制剂拉帕替尼(lapatinib)、治疗胃肠道基质肿瘤和转移性肾细胞癌的酪氨酸激酶抑制剂舒尼替尼(sunitinib)等[10]。但该技术仍具有筛选成本较高、耗时较长、分子多样性受制于化合物筛选库、较难对某些复杂靶点进行筛选等缺点。目前各研发团队仍在积极创新HTS技术,并广泛应用于创新药物研发。根据待测样品的种类,HTS可分为分子水平筛选和细胞水平筛选两大类。分子水平的筛选主要是检测受体功能的改变、蛋白质结合的抑制以及受体-配体结合的结构、动力学和亲和力等,常见的检测方法有荧光法(荧光偏振、荧光共振能量转移、酶联免疫吸附等)和非荧光法[表面等离子体共振(surface plasmon resonance,SPR)、核磁共振(nuclear magnetic resonance,NMR)、质谱分析(mass spectrum,MS)等]。中国医学科学院医药生物技术研究所联合皖南医学院开发一种基于荧光偏振和生物素-亲和素反应的新冠病毒主蛋白酶(Mpro)小分子抑制剂高通量筛选方法。研究团队合成一种异硫氰酸荧光素和生物素双标记的小分子肽(FITC-S-Biotin)作为荧光偏振示踪剂和Mpro的水解底物,活性化合物可以通过抑制Mpro对FITC-S-Biotin的水解作用,改变荧光强度。研究团队应用该模型从天然产物化合物库中筛选到新冠病毒Mpro竞争性抑制剂二鹅掌菜酚(Dieckol),半数抑制浓度(IC50)为(4.5±0.4) μmol·L-1[11]。上海科技大学研究团队建立一种高通量、无标签的亲和质谱筛选技术,用于筛选靶向G蛋白偶联受体(G protein-coupled receptors,GPCR)的小分子配体,通过对4333个化合物进行筛选,发现了1个5-羟色胺(5-ydroxytryptamine,5-HT)受体拮抗剂和4个胰高血糖素样肽-1受体(glucagon-like peptide-1receptor,GLP-1R)阳性变构调节剂[12]。细胞水平的筛选是在细胞个体水平完成的检测,常见的检测方法有离子通道检测、报告基因检测和细胞增殖检测,分别用于原发性电障碍等离子通道类疾病、瑞特综合征(rett syndrome,RTT)和阿尔兹海默病等脑部疾病、肿瘤和病毒感染等疾病的药物筛选[13]。中国科学院上海药物研究所研究人员利用表达钾离子通道蛋白家族KCNQ2的仓鼠卵巢细胞(CHO),建立一种改进的HTS方法,通过铊通量测定法从80000个化合物中筛选出565个比阳性化合物活性更强的KCNQ2通道激动剂,然后使用384孔自动化膜片钳和传统膜片钳,筛选得到ZG1732和ZG208,半数有效浓度(EC50)分别为(31.04±0.18),(1.37±0.06) μmol·L-1[14]。麻省理工学院怀特黑德生物医学研究所研究人员使用CRISPR-Cas9基因编辑技术,将荧光素酶报告基因插入人胚胎干细胞内源性K+/Cl-共转运蛋白2(KCC2)基因位点中,通过检测荧光强度,从900个化合物中筛选得到14个KCC2表达增强化合物(KEECs),有望用于RTT的治疗[15]。美国默克公司研究团队以β干扰素(IFN-β)的分泌为表型,在单核细胞系THP-1中进行高通量筛选,发现口服非核苷酸干扰素基因刺激因子(stimulator of interferon genes,STING)激动剂MSA-2,能够以二聚体形式结合并激活STING,靶向肿瘤组织发挥持久高效的抗肿瘤免疫活性[16]。2.2 SBDDSBDD技术起源于20世纪末,是指从配体和靶点的三维结构出发,以分子识别为基础而进行的药物设计方法,主要目的是预测与靶点结合位点产生最佳相互作用的化合物,可分为基于受体结构的药物设计和基于配体结构的药物设计两大类。其中,基于受体结构的药物设计是根据受体大分子的三维结构,通过计算机辅助药物设计(computer aided drug design,CADD)等方法,确定小分子与受体的结合构象,评价结合活性,筛选出有潜力的配体小分子; 基于配体结构的药物设计是依据现有药物的结构、理化性质与活性关系的分析,建立定量构效关系或药效基团模型,设计新的化合物或具有新骨架的活性分子[17]。1995年,基于该策略有2个药物首次获得美国食品药品管理局(FDA)批准,分别为用于降低开角型青光眼和高眼压症眼压增高的碳酸酐酶抑制剂多佐胺、治疗艾滋病的人类免疫缺陷病毒(human immunodeficiency virus,HIV)蛋白酶抑制剂沙奎那韦[18]。在CADD等技术的辅助下,SBDD显著提高了药物筛选命中率,具有开发成本较低、可从少量化合物筛选获得先导化合物、可直接预测受体和配体结合能力等优点。截至目前,FDA批准的药物大多基于该技术演化而来。但该技术需要受体完整的三维立体结构,且仅考虑受体和配体的结合强度而不能预测药效。随着结构生物学、人工智能与深度学习等不断突破,SBDD也在快速发展。麻省理工学院研发团队使用含有2335个已知抗菌活性的分子集合训练深度神经网络,该算法无需对药物进行标记,就可以分析化合物的分子结构并筛选潜在的抗生素。研究人员利用该算法筛选出的抗生素Halicin,与传统抗生素结构不同,显示出对包括结核分枝杆菌和碳青霉烯类耐药肠杆菌科在内的广泛病原菌系统发育谱的杀菌活性,并能有效治疗小鼠模型中难辨梭状芽孢杆菌和泛耐药鲍曼不动杆菌感染。Halicin是首次在没有任何人为假设的前提下,从零开始发现的全新抗生素[19]。印度TCS公司研发团队利用深度学习开发一种由图注意力网络和递归神经网络组合形成的条件生成模型,该模型利用图注意力网络学习活性位点的残基结构和相互作用,在药物靶向亲和力预测模型的指导下产生特定于靶向活性位点的小分子。研究人员在Janus激酶2(JAK2)和多巴胺受体D2(DRD2)上验证该方法,生成类似已知蛋白抑制剂的分子[20]。2.3 FBDD1981年JENCKS等[21]提出FBDD技术的概念和理论框架,认为某个类药性分子可以视为两个或多个具有生物活性小分子碎片的叠加。FBDD是利用NMR、SPR、X-射线单晶衍射(X-ray)以及TSAs等方法筛选出与靶蛋白具有相互作用的小分子弱活性片段,然后基于其结构信息对活性片段进行优化,得到具有更高活性的先导化合物的方法。该技术主要包括片段库的构建、活性小片段的筛选、片段的结构优化等步骤。其中,在构建片段库阶段,ERLANSON等[22]在先导化合物设计类药“五原则”的基础上,提出构建片段库的“三法则”,即片段的分子量<300,脂水分配系数<3,氢键供体与受体的数量分别<3。除此之外,片段库的大小需要根据所选择的筛选方法进行考虑,例如采用NMR或X-ray时,片段库的大小通常为1×102~1×103; 采用SPR时,因其具有高通量的特点,片段库大小可以达到1×105。片段的筛选是FBDD技术的核心,由于片段与靶点的结合作用较弱,因此需要高灵敏度和高稳定性的筛选方法检测,以满足所需的灵敏度和稳定性。随着技术的发展,微量热泳动、热迁移分析(TSAs)、弱亲和色谱等检测手段对NMR、SPR、X-ray等经典方法进行补充。通常经过筛选得到的初始片段活性很低,所以筛选后的关键是通过片段自组、片段连接或片段生长对片段进行结构优化,改善片段对靶标的选择性、生物利用度、生物转化等性质,使之成为候选药物,这也是FBDD技术最具挑战的环节[22]。与HTS比较,FBDD有以下优点,一是收集、维护和筛选片段库比化合物库更加容易,且筛选包含几千个片段的片段库就可以达到筛选化合物库的效果,研发周期更短; 二是具有更高的筛选命中率,可以实现对复杂靶标尤其涉及蛋白-蛋白相互作用靶点的处理; 三是片段的尺寸小、溶解度高,通常具有更好的药物属性,后期易于结构优化,有潜力成为活性高、选择性高的药物分子。截至目前,共有4个获批上市的药物利用FBDD技术筛选得到,分别为治疗黑色素瘤的丝氨酸/苏氨酸蛋白激酶B-raf(BRAF)抑制剂维莫非尼(vemurafenib)、治疗慢性淋巴细胞白血病的B细胞淋巴瘤因子2(Bcl-2)抑制剂维奈克拉(venetoclax)、治疗尿路上皮癌的成纤维细胞生长因子受体(fibroblast growth factor receptor,FGFR)抑制剂厄达替尼(erdafitinib)、治疗腱鞘巨细胞瘤的集落刺激因子1/干细胞因子受体(CSF1R/cKit)抑制剂Pexidartinib,其中维莫非尼从片段筛选到获批上市仅用6年时间[23-24]。该技术同样具有一定的局限性,如技术门槛要求较高,需要有较好的片段储备,且很大程度依赖于靶蛋白的三维结构信息,对纯化蛋白的需求量较大,片段经过结构优化后得到的先导化合物可能与片段分子结合位点不同,对筛选所需检测手段的灵敏度要求较高。各研发团队仍在积极开展相关研究工作。上海科技大学和复旦大学研究人员构建一种基于亲和质谱的FBDD筛选技术,与基于SPR或NMR的FBDD比较,该技术通过使用亲和质谱富集特定结合物,可减少2~4倍目标蛋白质和片段库化合物的使用量,分析速度可以提高2~3倍。研究人员利用该技术成功筛选到一种潜在的GPCR负变构调节剂Fg754,Fg754能够与钠离子变构位点特异性结合,且结合模式不同于已知的负变构调节剂[25]。印度昌迪加尔医学教育研究所研究人员以新冠病毒Mpro为靶点,利用FBDD技术对一个含有约20万化合物片段的片段库进行筛选,并将任何与相邻子口袋具有高亲和力的片段进行连接,最终得到17个与Mpro关键结合位点具有较高结合活性的分子,为新冠病毒候选药物研发提供更多选择[26]。2.4 DELDEL技术是一项基于组合化学和DNA技术的药物筛选方法,于1992年由美国Sydney Brenner和Richard Lerner提出,主要包括DNA编码化合物库的构建、DEL筛选以及先导化合物的产生等步骤[27]。该技术首先将每个化合物与一段特定的DNA分子序列进行连接,进行DNA编码,用作可扩增的识别条形码,构建经DNA编码的化合物库; 然后将活性靶蛋白和经DNA编码的化合物库进行亲和筛选,洗脱除去与靶蛋白亲和力弱或不结合的化合物,得到亲和力强的化合物集合; 由于化合物与DNA编码信息一一对应,筛选完成后利用高通量测序对筛选出化合物连接的DNA序列进行识别,确定编码对应的化合物分子; 最后重新合成不带DNA标签的化合物进行活性验证及结构优化,得到先导化合物[28]。DEL技术使用DNA标签作为条形码,可构建和筛选规模达几百万至数十亿种化合物的化学文库,具有库容量巨大、分子多样性好、对靶标蛋白需求量少等优点,DEL技术并非传统的一对一筛选,是将靶蛋白和整个编码化合物库同时孵育,具有显著的时间和成本优势,筛选周期为3~6个月,每个化合物筛选成本平均0.002美元。但DEL技术要求较高,暂无利用该技术获批上市的药物,且大体积的DNA编码标签以及组合化学在一定程度上增加筛选的复杂性和不确定性,需要进一步开发和优化与DNA相容的化学反应以保持化合物的类药性和库的纯度。此外,该技术的筛选对象主要针对纯化的生物靶点,对于难以表达的靶点或者活细胞体系等功能性靶点筛选较为困难[28-29]。目前,DEL技术已被国内外制药公司广泛应用,成为筛选先导化合物的重要手段,国内外DEL技术较为成熟的公司有4家,分别为英国葛兰素史克公司(GSK)、美国X-Chem公司、成都先导公司和丹麦Nuevolution公司。其中,GSK是对DEL技术应用最为成熟的企业,DEL库达几十亿量级,分子库筛选的靶点种类繁多,几乎涵盖所有疾病类型,但其DEL库的化学结构类型只有一种,为三嗪类杂环化合物,其技术仅供自用。GSK已有3个在研药物处于Ⅱ期临床试验阶段,分别为用于治疗糖尿病或心脑血管疾病等的可溶性环氧化物水解酶抑制剂GSK2256294、用于治疗银屑病或类风湿关节炎以及溃疡性结肠炎的ATP竞争型受体相互作用蛋白1(RIP1)抑制剂GSK2982772、用于治疗胰腺癌的RIP1抑制剂GSK3145095,其中GSK2256294是第一个由DEL技术发现并进入临床试验的小分子化合物[30]。成都先导公司是国内首个开展DEL研究的制药公司,目前已建立一个基于DEL的早期药物开发平台,该DEL库分子数量已超过1.2万亿,合成分子骨架的种类超过6000种,已有3个在研药物进入临床试验阶段,分别为用于治疗骨髓瘤或实体瘤的组蛋白去乙酰化酶(HDAC)抑制剂HG146、用于治疗晚期实体瘤的STING激动剂HG381、用于治疗具有神经营养受体酪氨酸激酶(NTRK)或C-ROS原癌基因1酪氨酸激酶(ROS1)基因融合的原肌球蛋白受体激酶(TRK)抑制剂HG030。美国贝勒医学院和德克萨斯儿童医院研究人员致力于筛选人类蛋白溴域和末端外亚群(BET)中第一个溴域(BD1)的特异性抑制剂,BD1是癌症等疾病的潜在靶标。该团队利用DEL技术,在一个试管中同时对40亿个DNA编码分子进行筛选,得到对BD1具有高度选择性的化合物CDD-724,该化合物抑制BD1的能力约是抑制其他人类溴化结构域的2000倍[31]。2.5 PROTACPROTAC是将靶向蛋白募集到E3泛素连接酶进行泛素化标记,然后通过泛素-蛋白酶体系统(UPS)将其降解的新型药物分子体系的方法。UPS是细胞内蛋白质降解的主要途径,参与细胞内80%以上蛋白质的降解。该系统由泛素(Ub)、3种泛素化酶(泛素活化酶E1,泛素结合酶E2s,泛素连接酶E3s)、蛋白酶体及其底物蛋白质等构成,在ATP供能的情况下,蛋白通过一系列酶的作用,被标记上多泛素化并转移到蛋白酶体内进行降解。PROTAC就是利用UPS原理设计的一种双功能小分子,由3部分构成,中间的连接体(Linker)一端连接可靶向目标蛋白的配体,另一端连接E3泛素连接酶配体分子,利用UPS识别、结合并降解疾病相关的靶蛋白[32]。该技术是一种全新药物设计策略,理论上可以将任何过表达和突变的致病蛋白清除,达到治疗疾病的目的[33]。PROTAC的发展经历了第1代基于多肽片段的设计,到2008年开始的第2代小分子PROTAC设计,降解的靶蛋白包括甲硫氨酰氨肽酶2、雄激素受体、细胞视黄酸结合蛋白、雌激素受体、Tau微管相关蛋白、激酶类等,涉及的疾病包括癌症、类风湿疾病、神经退行性疾病等[32]。基于作用机制,PROTAC主要有以下优点: 一是PROTAC分子不直接抑制靶蛋白的功能活性,不需要与靶蛋白发生长时间和高强度的结合,可以靶向转录因子、支架蛋白和非酶蛋白等对于传统小分子药物“无成药性”的蛋白; 二是相比于传统小分子药物的“占位驱动”模型,PROTAC分子属于“事件驱动”,只需要瞬态结合就可以直接催化降解靶蛋白,因此使用催化剂量即可发挥药物疗效,耐药性更低; 三是由于靶蛋白与E3泛素连接酶之间的协同作用,PROTAC分子具有更高的选择性。尽管PROTAC技术从肽到全小分子有了显著的提升,但是与传统的小分子药物相比,PROTAC分子作为三元复合物,分子量较大,提高水溶性、稳定性、口服生物利用度、血管穿透能力等需进一步研究。此外,由于泛素化标记不仅涉及到蛋白质的降解,还关系到甲基化、乙酰化、磷酸化等过程以及DNA,其脱靶毒性仍是目前面临的难题[6,32]。PROTAC作为一种新兴的小分子药物研究领域,吸引了众多制药企业和学术机构开展研究。创立于2013年的美国Arvinas公司是该领域的领头羊,研发管线主要包括抗肿瘤药物和神经疾病药物,目前共有Bavdegalutamide(ARV-110)、ARV-471、ARV-766三款候选药物处于临床阶段。其中,Bavdegalutamide选择性靶向降解雄激素受体(androgen receptor,AR),主要用于治疗转移性趋势抵抗性前列腺癌(mCRPC),是全球首个进入临床试验的口服PRAOTC小分子药物,目前处于Ⅱ期临床试验阶段。2022年2月,Arvinas公布的Ⅱ期临床试验结果显示,Bavdegalutamide展现出持续抗肿瘤活性和患者获益证据,在携带AR T878X/H875Y突变的肿瘤患者中,可以使46%患者的前列腺特异性抗原(prostatic specific antigen,PSA)水平降低≥50%。ARV-471靶向雌激素受体(estrogen receptor,ER),用于治疗ER阳性/人表皮生长因子受体2(human epidermal growth factor receptor2,HER2)阴性(ER+/HER2-)的乳腺癌,于2022年12月启动Ⅲ期临床试验,是目前研究进展最快的PRAOTC分子。2022年11月,公布的Ⅱ期临床试验初步结果显示,ARV471具有良好的耐受性,显示出38%的临床获益率(CBR: 确认完全缓解率、确认部分缓解率或疾病稳定率>24周),对于ESR1突变患者,CBR为51.2%[34]。国内多家药企也在积极开展相关研究,如海思科公司的HSK29116、开拓药业的GT20029、百济神州公司的BGB-16673等均处于Ⅰ期临床试验阶段; 糺诺生物公司的RNK05047处于Ⅰ/Ⅱ期临床试验阶段,于2022年8月在美国完成首例患者给药。上述不同技术优劣势比较见表1。3展望随着生物技术的飞速发展,以重组蛋白质药物、治疗性抗体、基因治疗、干细胞治疗等为代表的生物技术药物成为新药研发的热点,小分子药物受到一定冲击,小分子药物分子类型和多样性的增速降低。但是,小分子药物作为最传统的药物形式,以其难以替代的优势,仍是药物研发领域的重要组成部分。通过对上述小分子药物筛选技术的发展现状分析可以发现,美国是该领域的领头羊,我国虽然在相关领域取得了一定进展,但仍落后于美国。因此,一是要以全面提升我国药物研发创新能力为导向,持续加强基础研究,积累原创性成果,提高源头创新力。二是要以更好的解决小分子药物研发过程中的共性问题为牵引,对于人工智能药物设计技术、智能药物制备技术、氘代药物开发技术等能够优化我国新药创制体系的关键共性技术,加强集中攻关,促进我国制药产业转型升级。三是要以直击现阶段小分子药物研发的痛点为目的,对于DEL、PROTAC、小分子辅助受体靶向技术等具有前瞻性、先导性和一定探索性的前沿引领技术,要提前优先布局,并不断将变革性新技术应用于小分子药物的研发,为生物医药产业发展注入新动力,为未来新药创制领域技术更新换代和新兴产业的发展奠定基础。参考文献《医药导报》 2024年2月 第43卷第2期文章信息源于公众号凡默谷,登载该文章目的为更广泛的传递行业信息,不代表赞同其观点或对其真实性负责。文章版权归原作者及原出处所有,文章内容仅供参考。本网拥有对此声明的最终解释权,若无意侵犯版权,请联系小编删除。学如逆水行舟,不进则退;心似平原走马,易放难收。行舟Drug每日更新 欢迎订阅+医药大数据|行业动态|政策解读
蛋白降解靶向嵌合体
100 项与 Nuevolution AB 相关的药物交易
登录后查看更多信息
100 项与 Nuevolution AB 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年07月09日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
其他
5
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
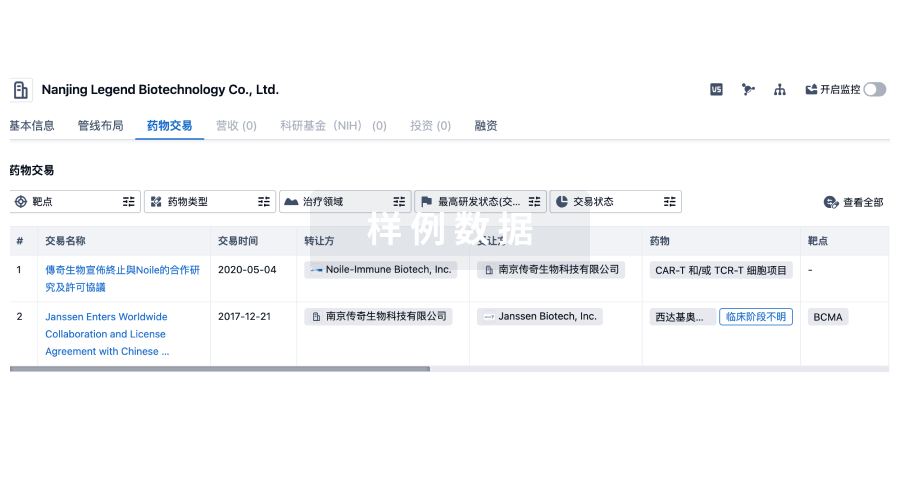
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
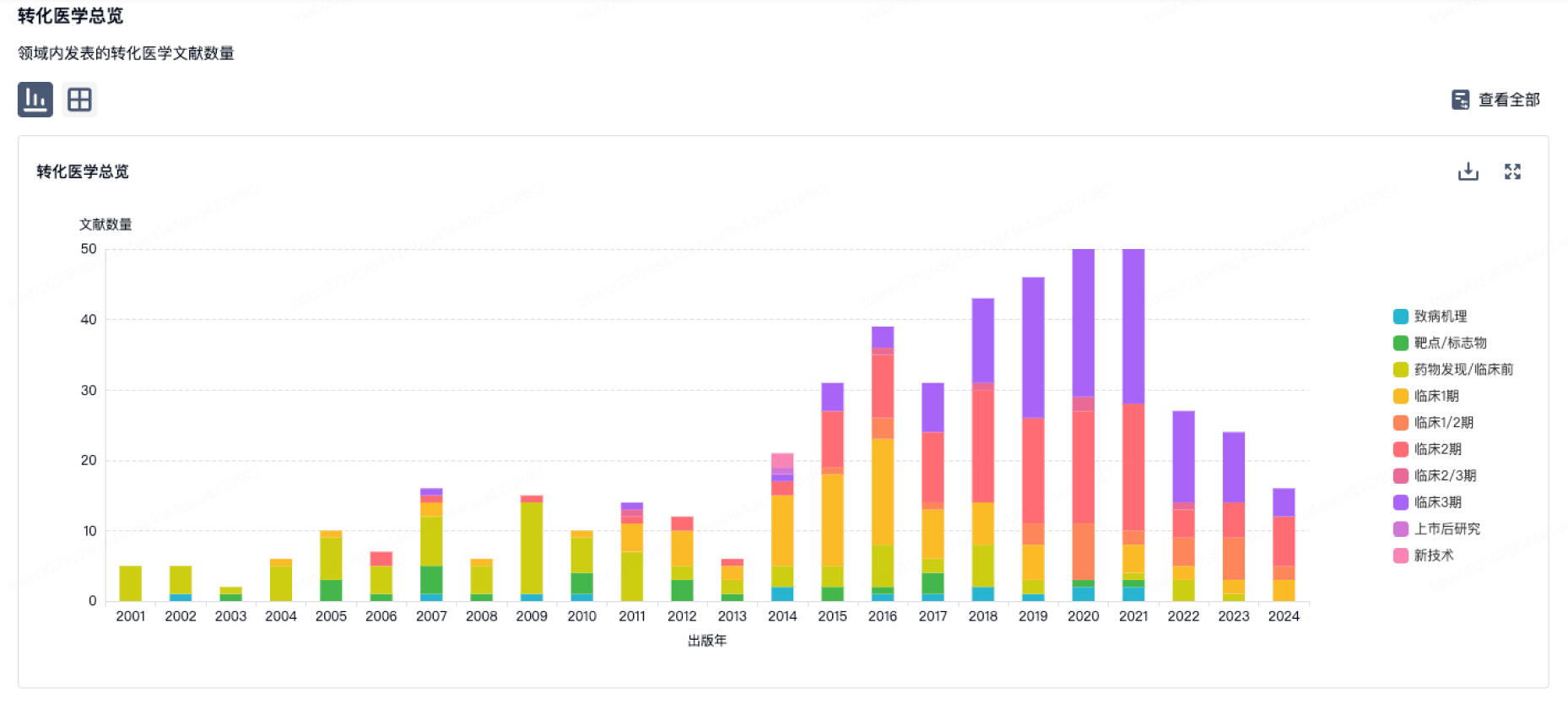
营收
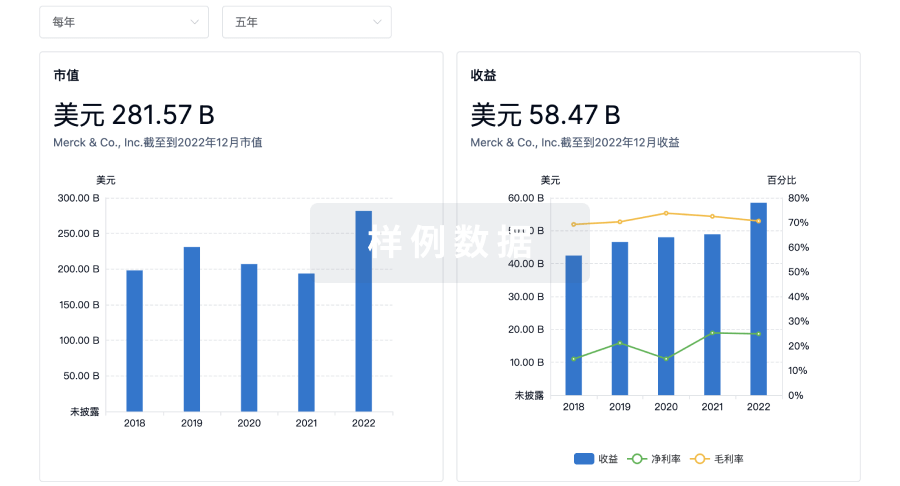
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
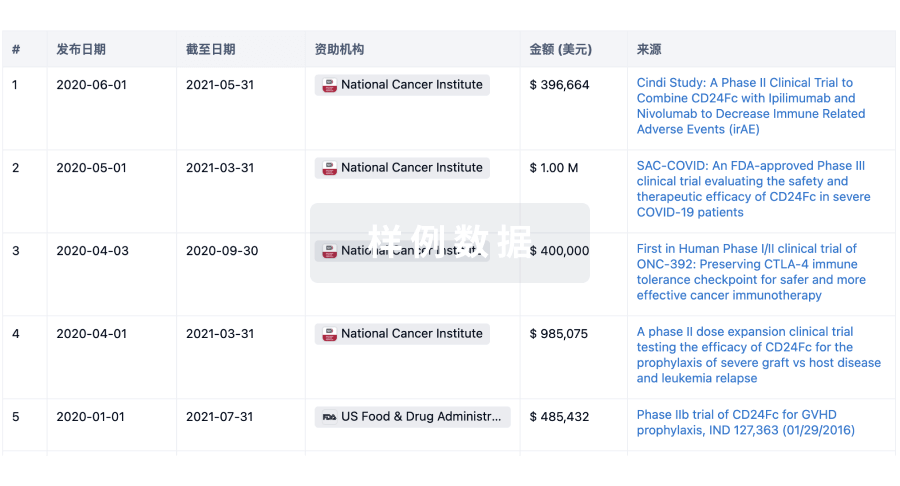
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用