预约演示
更新于:2026-03-03

Oxford Nanopore Technologies Plc
更新于:2026-03-03
概览
关联
100 项与 Oxford Nanopore Technologies Plc 相关的临床结果
登录后查看更多信息
0 项与 Oxford Nanopore Technologies Plc 相关的专利(医药)
登录后查看更多信息
20
项与 Oxford Nanopore Technologies Plc 相关的文献(医药)2021-12-01·GENOME BIOLOGY
Complete vertebrate mitogenomes reveal widespread repeats and gene duplications
Article
作者: Ambrosini, Roberto ; Torrance, James ; Fedrigo, Olivier ; Bista, Iliana ; Rhie, Arang ; Durbin, Richard ; Koren, Sergey ; Horner, David S ; Balacco, Jennifer ; Uliano-Silva, Marcela ; Achilli, Alessandro ; Wood, Jonathan ; Costa, Vania ; Betteridge, Emma ; Corton, Craig ; McCarthy, Shane ; Mayes, Simon ; Fungtammasan, Arkarachai ; Jarvis, Erich D ; Mountcastle, Jacquelyn ; Howe, Kerstin ; Dolucan, Jale ; Chiara, Matteo ; Capodiferro, Marco Rosario ; Tracey, Alan ; Winkler, Sylke ; Haase, Bettina ; Formenti, Giulio ; Myers, Eugene ; Brown, Samara ; Oliver, Karen ; Fordham, Daniel ; Smith, Michelle ; Skelton, Jason ; Phillippy, Adam M ; Al-Ajli, Farooq O ; Kwak, Woori ; Braun, Edward L ; Korlach, Jonas ; Houde, Peter
Abstract:
Background:
Modern sequencing technologies should make the assembly of the relatively small mitochondrial genomes an easy undertaking. However, few tools exist that address mitochondrial assembly directly.
Results:
As part of the Vertebrate Genomes Project (VGP) we develop mitoVGP, a fully automated pipeline for similarity-based identification of mitochondrial reads and de novo assembly of mitochondrial genomes that incorporates both long (> 10 kbp, PacBio or Nanopore) and short (100–300 bp, Illumina) reads. Our pipeline leads to successful complete mitogenome assemblies of 100 vertebrate species of the VGP. We observe that tissue type and library size selection have considerable impact on mitogenome sequencing and assembly. Comparing our assemblies to purportedly complete reference mitogenomes based on short-read sequencing, we identify errors, missing sequences, and incomplete genes in those references, particularly in repetitive regions. Our assemblies also identify novel gene region duplications. The presence of repeats and duplications in over half of the species herein assembled indicates that their occurrence is a principle of mitochondrial structure rather than an exception, shedding new light on mitochondrial genome evolution and organization.
Conclusions:
Our results indicate that even in the “simple” case of vertebrate mitogenomes the completeness of many currently available reference sequences can be further improved, and caution should be exercised before claiming the complete assembly of a mitogenome, particularly from short reads alone.
2021-09-01·Biomaterials1区 · 工程技术
Detection of single peptide with only one amino acid modification via electronic fingerprinting using reengineered durable channel of Phi29 DNA packaging motor
1区 · 工程技术
Article
作者: Zhang, Long ; Jordan, Michael ; Jayasinghe, Lakmal ; Burns, Nicolas ; Freitas, Michael A ; Aldana, Julian ; Gardner, Miranda L ; Guo, Peixuan
Protein post-translational modification (PTM) is crucial to modulate protein interactions and activity in various biological processes. Emerging evidence has revealed PTM patterns participate in the pathology onset and progression of various diseases. Current PTM identification relies mainly on mass spectrometry-based approaches that limit the assessment to the entire protein population in question. Here we report a label-free method for the detection of the single peptide with only one amino acid modification via electronic fingerprinting using reengineered durable channel of phi29 DNA packaging motor, which bears the deletion of 25-amino acids (AA) at the C-terminus or 17-AA at the internal loop of the channel. The mutant channels were used to detect propionylation modification via single-molecule fingerprinting in either the traditional patch-clamp or the portable MinION™ platform of Oxford Nanopore Technologies. Up to 2000 channels are available in the MinION™ Flow Cells. The current signatures and dwell time of individual channels were identified. Peptides with only one propionylation were differentiated. Excitingly, identification of single or multiple modifications on the MinION™ system was achieved. The successful application of PTM differentiation on the MinION™ system represents a significant advance towards developing a label-free and high-throughput detection platform utilizing nanopores for clinical diagnosis based on PTM.
2021-07-01·Nature chemistry1区 · 化学
Constructing ion channels from water-soluble α-helical barrels
1区 · 化学
Article
作者: Thomson, Andrew R ; Brady, R Leo ; Liu, Lijun ; Kratochvil, Huong T ; DeGrado, William F ; Sengel, Jason T ; Wallace, Mark I ; Woolfson, Derek N ; Scott, Alistair J ; Bayley, Hagan ; Mulholland, Adrian J ; Niitsu, Ai ; Dawson, William M ; Mahendran, Kozhinjampara R ; Mravic, Marco ; Lang, Eric J M
The design of peptides that assemble in membranes to form functional ion channels is challenging. Specifically, hydrophobic interactions must be designed between the peptides and at the peptide-lipid interfaces simultaneously. Here, we take a multi-step approach towards this problem. First, we use rational de novo design to generate water-soluble α-helical barrels with polar interiors, and confirm their structures using high-resolution X-ray crystallography. These α-helical barrels have water-filled lumens like those of transmembrane channels. Next, we modify the sequences to facilitate their insertion into lipid bilayers. Single-channel electrical recordings and fluorescent imaging of the peptides in membranes show monodisperse, cation-selective channels of unitary conductance. Surprisingly, however, an X-ray structure solved from the lipidic cubic phase for one peptide reveals an alternative state with tightly packed helices and a constricted channel. To reconcile these observations, we perform computational analyses to compare the properties of possible different states of the peptide.
136
项与 Oxford Nanopore Technologies Plc 相关的新闻(医药)2026-03-01
·循因缉药
全球视野,深度视角
各位亲爱的股东,大家早上好中午好晚上好!
我真是服了...手里还攥着一堆财报分析...不过,吃瓜最重要
PART.01
“不正常”
我们先看图片。
翻译如下(by Kimi,人工微调):
Oxford Nanopore Technologies plc公司今日表示,在针对MGI澳大利亚有限公司等(以下简称"MGI")的澳大利亚专利诉讼中,MGI已承认其"Cyclone SEQ WT02"产品侵犯了牛津纳米孔的四项澳大利亚专利。
杰克曼法官在最近的案件管理听证会上确认,侵权责任已无争议。审判定于2027年进行,届时将对MGI剩余的抗辩理由进行裁决。关于额外或加重赔偿的审议将在审判结束后进行。
另在英国,牛津纳米孔已向高等法院提起诉讼,指控多家MGI/华大基因实体侵犯商业秘密、违反保密义务及违反合同。
在这两起案件中,牛津纳米孔均提起法律诉讼以保护支撑其传感平台的知识产权(以下简称"IP"),这些权利已获得澳大利亚及许多其他司法管辖区的专利授权。该公司持续投入创新,并致力于保护这一知识产权,以此作为为全球客户和合作伙伴创造长期价值的核心驱动力。
经常打专利官司的朋友们肯定会嗅到不同寻常的味道,通常情况下只要被指控侵权的公司经营正常,是绝对不会在审前直接“滑跪”的。
所以在看到ONT官方提交的这份公开文件那一刻我们就绷不住了,首先想到的竟然是MGI澳大利亚分部“叛变”了...
另外,对于“Justice Jackman confirmed at a recent case management hearing that liability for infringement is not in dispute.”这段话我们也是持有怀疑态度的。
这里缺乏明确的指向,这玩意甚至可能只是说对于控方指控的内容没有争议。
相信我,对于英国人的语言艺术打过交道的都懂。
那么,真相是什么?
PART.02
“反击”
MGI也迅速做出了回应,甚至我感觉这个网站上的排版都没来得及好好搞搞,不过无伤大雅。
翻译如下(by Kimi,人工微调):
我们注意到牛津纳米孔技术公司(Oxford Nanopore Technologies plc)就澳大利亚正在进行的知识产权诉讼发表的公开声明。
需要明确的是,该案件定于2027年6月进行审判,目前仍在审理中,尚未有任何法院就案件实质作出判决。我们已主张涉案专利因各种原因无效,应予以撤销。我们对自身的法律立场充满信心,并将继续通过适当的法律渠道维护我们的权利和利益。
我们认为尊重法律程序至关重要。任何暗示案件已有定论的表述都为时过早,并不能反映案件的程序性现状。我们强烈反对任何可能误导公众、歪曲案件程序性现状的表述,尤其是在案件仍在审理期间。我们敦促各方确保公开声明准确反映正在进行的司法程序。
关于据报道已在英国提起的诉讼,我们尚未收到任何正式送达的诉状。
我来给大家中译中翻译一下标出的这3个点:
第一,MGI认为ONT所谓的专利无效,根本就不存在专利侵权,更谈不上承认侵犯。
专利有效是侵权发生的前提,你都无效了,当然就失去了一切的基础。
第二,不要半场开香槟,我是尊重法律程序的,你遵不遵守大家现在都看见了。
Emmm,有水平,语言的艺术到位了。
第三,警告你,不要搞“舆论战”。
我甚至觉得ONT这波是被“赢学传染”入脑了...
PART.03
“赢学传染”
当年,2024年9月9日CycloneSeq WT-02刚一发布,ONT就按捺不住在美国对华大提起了诉讼。
结果呢?2024年11月4日,ONT撤回了传票申请。
在发布会次日就发起诉讼,从商业考量上无可厚非。
大家都是做生意,法律允许前提下玩多脏都是你的本事。
但是这次性质就有点不一样了,有点太心急了啊,就想着迫切宣布一场胜利来证明自己的孩子。
这让我想起了一位故人...“赢学”的风还是吹到了不列颠...
其实也好理解,不仅仅有中国MGI华大智造的CycloneSeq纳米孔测序仪,ONT还要面对Roche Axelios迫在眉睫的威胁。
想赢,正常。
PART.04
最后
我们来说下专利纠纷的一点点常识:
在没有极度特殊压力的情况下,被控侵权方不会直接承认侵权事实。
这跟你在家偷偷看电视,大人回家发现电视热的怀疑你时直接承认一样,既不能避免挨揍还显得自己没骨气。
什么早早承认侵权可以避免消耗战更是扯淡,要记住,一般企业的专利布局不是一项而是一个体系。
这次,他可以用其中4个专利搞你,下次就可以用其他5个专利搞你。
最后的最后,专利官司是长期斗争,且不到最后一刻,永远存在变数,千万别被“赢学”宣传误导。
好了,明天的瓜明天再吃,散会!
Tags:#纳米孔测序 #ONT #MGI #CycloneSeq #华大智造 #专利
END
各位股东看了么?赞了么?转发了么?别忘了点赞。谢谢!
所有内容均不作为投资建议,信息均来自公开资料,星球更新更快。
扫码加入星球!
循因缉药Elite
PS:风里雨里,老编等你~
近期原创:
Roche纳米孔测序仪开卖!每M Reads 6分钱!150美元的基因组?!
安捷伦Agilent 2026Q1:亚洲强劲增长!中国区+6%,超出预期!
测序仪回旋镖:“你们只会堆通量吗?”
Bruker布鲁克:中国区的反弹结束了
Q70!提速30%!Illumina“杀死了比赛”??
相关资料:
注1:https://otp.tools.investis.com/clients/uk/oxford_nanopore/rns/regulatory-story.aspx?cid=2700&newsid=2037029
注2:https://global-mgitech.com/mgi-statement-regarding-ongoing-intellectual-property-litigation-in-australia/
专利侵权财报
2026-01-28
WOIPPY, FRANCE, January 28, 2026 /
EINPresswire.com
/ -- ABL Diagnostics (FR001400AHX6 – “ABLD”), an innovative company specialized in microbiology and pathogen sequencing, announces the reinforcement of its strategic agreements with leading global life science and molecular biology suppliers, including Thermo Fisher Scientific, QIAGEN, Vazyme, Integrated DNA Technologies (IDT), Illumina, Roche, Biolegio, Eurofins, Oxford Nanopore Technologies, MGI, and other key industry partners.
These strengthened partnerships reflect ABL Diagnostics’ commitment to securing a robust, reliable, and scalable supply chain capable of supporting the strong growth of its commercial activities across all business lines.
The renewed and expanded agreements provide improved commercial conditions, including more competitive pricing, optimized contractual terms, and enhanced supply security. These improvements will enable ABL Diagnostics to better manage increasing volumes while maintaining high standards of quality and performance.
The reinforced supplier framework directly supports ABL Diagnostics’ core technologies, notably genotyping by sequencing and qPCR-based molecular detection, both of which are experiencing sustained demand across research and diagnostic markets.
By benefiting from improved tariffs and more favorable terms, ABL Diagnostics strengthens its cost structure and operational efficiency. These conditions translate into improved margin expectations and enhanced growth visibility, positioning the company to meet its ambitious commercial objectives.
As a result, ABL Diagnostics enters 2026 with reinforced financial and growth forecasts, supported by long-term partnerships with key industry players and a supply ecosystem aligned with its expansion strategy.
This milestone confirms ABL Diagnostics’ position as a fast-growing and reliable partner in molecular diagnostics, fully equipped to respond to increasing market demand and to sustain long-term value creation.
***
About ABL Diagnostics (ABLD)
ABL Diagnostics (ABLD) is an international company that specializes in innovative molecular biology tests and global solutions for its customers:
- Molecular polymerase chain reaction (PCR) detection – UltraGene, and
- Genotyping by DNA sequencing – DeepChek
®
.
ABL Diagnostics markets its entire product range globally through its own sales team and a network of exclusive distributors active on all continents. ABL Diagnostics' customers are academic clinical pathology laboratories, private reference laboratories and researchers willing to implement innovative and robust microbiological content in constant expansion.
ABL Diagnostics has been marketing the products and services of its sister company CDL Pharma since the second half of 2025 through an intra-group strategy agreement.
An expanding portfolio of microbiology products:
- HIV – Drug resistance testing, including a whole genome kit.
- SARS-CoV-2, Tuberculosis, Hepatitis B and C – Advanced Detection Solutions.
- Microbiome and taxonomy – 16s/18s RNA-based analyses.
- Other viral and bacterial targets – Comprehensive molecular assays.
Integrated Solutions
- Real-time syndromic PCR tests
- Nadis
®
– Patient Medical Record used in more than 200 hospitals in France for the management of HIV and hepatitis.
- MediaChek
®
– Clinical Sample Collection Kits.
ABL Diagnostics, headquartered in Woippy, is a public limited company listed on compartment B of the regulated market of Euronext in Paris (Euronext: ABLD – ISIN: FR001400AHX6). These molecular biology products generate recurring revenues and cover one of the largest portfolios of applications in microbiology.
Contact
ABL Diagnostics SA
Société anonyme au capital de 1 611 465,60 euros
Headquarters : 72C route de Thionville - 57140 WOIPPY
552 064 933 R.C.S. METZ Tel : +33 (0)7 83 64 68 50
Email : info@abldiagnostics.com
https://www.abldiagnostics.com/
Dr Sayada
ABL Diagnostics SA
+33 7 83 64 68 50
email us here
Legal Disclaimer:
EIN Presswire provides this news content "as is" without warranty of any kind. We do not accept any responsibility or liability
for the accuracy, content, images, videos, licenses, completeness, legality, or reliability of the information contained in this
article. If you have any complaints or copyright issues related to this article, kindly contact the author above.
诊断试剂
2026-01-22
DELRAY BEACH, Fla., Jan. 22, 2026 /PRNewswire/ -- According to MarketsandMarkets™, the
Next-Generation Sequencing Market is projected to grow from about USD 14.94 billion in 2025 to USD 29.53 billion by 2030, at a CAGR of 14.6%.
Browse 1,100 market data tables and 65 figures spread through 720 pages and in-depth TOC on "NGS market- Global Forecast to 2030"
Next-Generation Sequencing Market Size & Forecast:
Market Size Available for Years: 2024–2030
2025 Market Size: USD 14.94 billion
2030 Projected Market Size: USD 29.53 billion
CAGR (2025–2030): 14.6%
Next-generation Sequencing Market Trends & Insights:
The next-generation sequencing market services segment is expected to register the highest CAGR of 18.2%.
In the next-generation sequencing market, by technology, the sequencing by synthesis segment will grow at a CAGR of 9.2%.
In the next-generation sequencing market, by application, the diagnostics segment is projected to grow at the fastest rate (13.9%) from 2025 to 2030.
The North American next-generation sequencing market accounted for a 41.8% revenue share in 2024.
Download PDF Brochure @
The main factors behind the expansion of this market are increased government funding for sequencing projects, rising applications of NGS in chronic disorders, and growing applications of NGS in clinical diagnostics.
Steady advances in bioinformatics and data analytics are key drivers of the NGS market's growth. These tools facilitate the processing of sequencing data and the extraction of valuable insights. Concurrently, a broader range of applications is becoming accessible as sequencing costs decline. Personalized medicine and genome research are also gaining momentum, driven by growing government funding and initiatives. Demand for sequencing solutions is rising as population genomics and large-scale sequencing initiatives provide deeper insights into genetic diversity and disease risk. Additionally, AI- and ML-enabled workflows are increasing efficiency and accuracy, accelerating innovation.
By
offering, the products segment accounted for the largest share in the next-generation sequencing market in 2024
The NGS market is segmented into products and services. The products segment accounted for the largest share of the NGS market in 2024. The products segment leads largely due to steady, recurring revenue from consumables, while the high price of NGS instruments also adds meaningful value to product sales. Services remain the second-largest segment in the market.
Request Sample Pages@
By platform technology, the sequencing by synthesis technology segment, accounted for the largest share in terms in 2024
The next-generation sequencing market is segmented into sequencing by synthesis, ion semiconductor sequencing, single-molecule real-time sequencing, nanopore sequencing, and other sequencing technologies. The segment comprising sequencing by synthesis constituted the largest share in 2024. This dominant share is attributed to the extensive use of the technique in genomics, transcriptomics, epigenomics, and metagenomics, thereby increasing its market penetration. The segment comprising ion semiconductor sequencing technologies held the second-largest share.
By service type, the products segment accounted for the largest share in the next-generation sequencing market in 2024
The next-generation sequencing market is segmented into sequencing services, presequencing services, bioinformatics and data analysis services, and NGS platform services. Sequencing services held the largest share in 2024, driven by large-scale genomics studies and programs run by governments and private organizations that often rely on established providers for high-throughput needs. Bioinformatics and data analysis services ranked second, supported by the growing volume and complexity of sequencing data.
By application, the diagnostics segment accounted for the largest share of the application segment in 2024.
The next-generation sequencing market is segmented into drug discovery & development, diagnostics, agriculture and animal research, and other applications. In 2023, the diagnostics segment accounted for the largest share of the NGS market. This can be attributed to declining sequencing costs, which have made NGS more accessible for routine diagnostic use in clinical labs and hospitals. Support for large-scale screening programs, such as newborn screening and population genomics projects, has also driven demand for NGS diagnostics.
By end user, the academic & research institutes segment accounted for the largest share of the product in the next-generation sequencing market in 2024.
The next-generation sequencing market is segmented into academic and research institutes, pharmaceutical and biotechnology companies, clinical and diagnostic laboratories, and other end users. Academic and research institutes held the largest share of the next-generation sequencing market in 2024. This is largely because NGS workflows are flexible and fit a wide range of custom study designs, aligning well with academic research needs. These institutes also lead early adoption of emerging areas such as single-cell sequencing and spatial transcriptomics, which depend on advanced NGS platforms and high consumable usage.
The North American region accounted for the largest share of the NGS market in 2024.
The NGS market is segmented into six major regions, namely North America, Europe, Asia Pacific, Latin America, the Middle East, and Africa. North America is the largest regional market for NGS in 2024, while Europe is the second largest.
The large share of North America can be attributed to the region's well-developed healthcare and research infrastructure, which supports the adoption of advanced NGS technologies in clinical and research settings. However, the Asia-Pacific region is estimated to be the fastest-growing segment of the market, owing to infrastructure development in developing countries, large-scale population genomics projects, and expanding collaborations among pharma and biotech companies for NGS testing and analysis.
Inquire Before Buying@
Top Companies in Next Generation Sequencing Market
:
The Top Companies in Next Generation Sequencing Market include Illumina, Inc. (US), Thermo Fisher Scientific Inc. (US), F. Hoffmann-La Roche Ltd. (Switzerland), Danaher Corporation (US), QIAGEN (Netherlands), Agilent Technologies, Inc. (US), Revvity (US), Eurofins Scientific (Luxembourg), PacBio (US), Oxford Nanopore Technologies plc. (UK), Takara Bio Inc. (Japan), BGI Group (China), Merck KGaA (Germany), BD (US), 10X Genomics (US), New England Biolabs (US), Promega Corporation (US), Novogene Co., Ltd. (China), LGC Limited (UK), WuXi Biologics (China), MGI Tech Co. Ltd. (China), Tecan Trading AG (Switzerland), Twist Biosciences (US), Azenta US, Inc. (US), GenScript (US), SD Biosensor, Inc. (South Korea), Fulgent Genetics (US), Hamilton Company (US), Zymo Research Corporation (US), NeoGenomics Laboratories (US), and Psomagen (US).
Browse Adjacent Markets:
Biotechnology Market Research
Reports & Consulting
Related Reports:
Single Cell Analysis Market
- Global Forecast to 2030
Life Science Instrumentation Market
- Global Forecast to 2031
Genomics Market
- Global Forecast to 2030
Drug Discovery Services Market
- Global Forecast to 2030
Molecular Diagnostics Market
- Global Forecast to 2030
About MarketsandMarkets™
MarketsandMarkets™ has been recognized as one of America's Best Management Consulting Firms by Forbes, as per their recent report.
MarketsandMarkets™ is a blue ocean alternative in growth consulting and program management, leveraging a man-machine offering to drive supernormal growth for progressive organizations in the B2B space. With the widest lens on emerging technologies, we are proficient in co-creating supernormal growth for clients across the globe.
Today, 80% of Fortune 2000 companies rely on MarketsandMarkets, and 90 of the top 100 companies in each sector trust us to accelerate their revenue growth. With a global clientele of over 13,000 organizations, we help businesses thrive in a disruptive ecosystem.
The B2B economy is witnessing the emergence of $25 trillion in new revenue streams that are replacing existing ones within this decade. We work with clients on growth programs, helping them monetize this $25 trillion opportunity through our service lines – TAM Expansion, Go-to-Market (GTM) Strategy to Execution, Market Share Gain, Account Enablement, and Thought Leadership Marketing.
Built on the 'GIVE Growth' principle, we collaborate with several Forbes Global 2000 B2B companies to keep them future-ready. Our insights and strategies are powered by industry experts, cutting-edge AI, and our Market Intelligence Cloud, KnowledgeStore™, which integrates research and provides ecosystem-wide visibility into revenue shifts.
To find out more, visit ™.com or follow us on Twitter , LinkedIn and Facebook .
Contact:
Mr. Rohan Salgarkar
MarketsandMarkets™ INC.
1615 South Congress Ave.
Suite 103, Delray Beach, FL 33445
USA: +1-888-600-6441
Email: [email protected]
Visit Our Website:
Logo:
SOURCE MarketsandMarkets
21%
more press release views with
Request a Demo
100 项与 Oxford Nanopore Technologies Plc 相关的药物交易
登录后查看更多信息
100 项与 Oxford Nanopore Technologies Plc 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月21日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
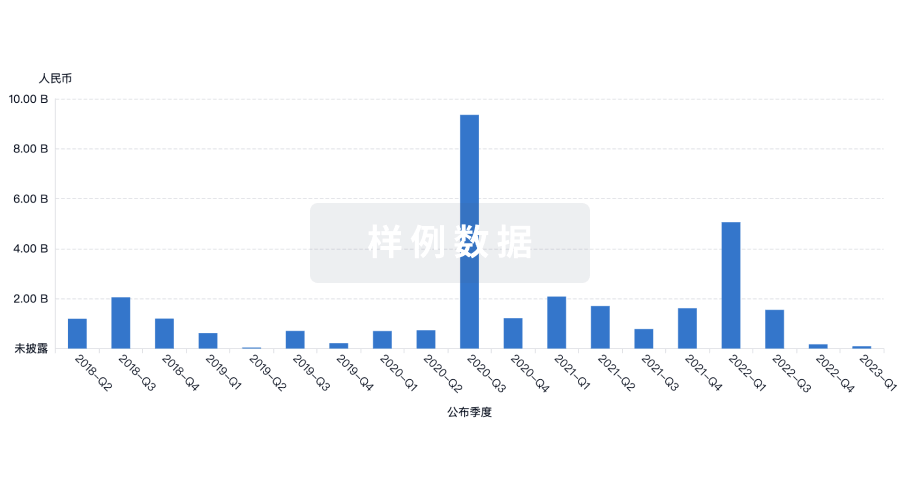
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用