预约演示
更新于:2025-05-07

Allied Scientific Products
更新于:2025-05-07
概览
关联
100 项与 Allied Scientific Products 相关的临床结果
登录后查看更多信息
0 项与 Allied Scientific Products 相关的专利(医药)
登录后查看更多信息
292
项与 Allied Scientific Products 相关的新闻(医药)2025-04-17
·药时空
近日,重组人血白蛋白领域有两则重磅新闻:一是安睿特重组人白蛋白注射液Ⅲ期临床试验完成揭盲,二是央视新闻一则“稻米造血”报道引起了广泛关注。01 重组人血白蛋白III期临床相继取得成功2025年4月16日,通化安睿特发布消息称,公司自主研发的重组人白蛋白注射液国内Ⅲ期临床研究完成揭盲,取得了良好的数据,其中包括完全符合国家要求的临床疗效指标和安全性数据。安睿特通过基因重组技术生产的重组人白蛋白,利用先进的酵母表达系统,实现了高纯度、高稳定性的超大规模生产,不仅规避了血浆来源限制,还显著降低了病毒污染风险,其分子结构与天然人血白蛋白高度一致。此次Ⅲ期临床研究采取多中心、随机、双盲、阳性药物对照设计,以治疗后血清ALB的变化为主要终点指标,比较重组人白蛋白(rHA)与人血白蛋白(HSA)注射液在肝硬化腹水患者低白蛋白血症中的疗效等效性,以腹水深度改善率作为关键次要指标比较非劣效性。初步结果显示:重组人白蛋白注射液治疗肝硬化腹水患者低白蛋白血症具有良好的疗效,与人血白蛋白疗效相当,起效速度相近且疗效维持时间更具优势,安全性和耐受性良好。详细结果将于近期公布。此前,通化安睿特的重组人白蛋白注射液已于2024年4月正式获得欧亚经济联盟—俄罗斯联邦卫生部批准上市,成为全球唯一上市销售的重组人白蛋白注射液产品,填补了全球生物医药产业的空白。除俄罗斯外,安睿特也与土耳其、乌兹别克斯坦等“一带一路”的多个国家达成了合作,同时与奥地利、墨西哥、阿尔及利亚、印度尼西亚等国家建立了广泛的伙伴关系,并计划在美国进行FDA快速临床申请。近期,通化安睿特的重组人白蛋白注射液还获得吉尔吉斯斯坦国家药品和医疗器械监督管理局颁发的MAH许可,这是继俄罗斯之后安睿特在海外获得的第二个MAH许可证;公司还与沙特阿拉伯Elaj Group签署了框架合作备忘录;与埃及医疗保健局就相关合作事宜正在进行探讨等。除通化安睿特外,禾元生物也在重组人血白蛋白的研发方面取得了重大进展。2024年11月底,禾元生物向全球公开发布了一项重大研究成果:其自主研发的重组人血清白蛋白注射液(奥福民,HY1001),在确证性临床研究中成功达到研究终点。禾元生物运用基因编辑技术,将人血清白蛋白基因精确植入水稻基因组,成功表达人血清白蛋白。数据显示,改造后的基因工程水稻,1公斤稻米产出约10克血清白蛋白,远超传统血浆提取技术的生产效率。因此,其又被称为“稻米造血”。奥福民已于2024年8月被国家药品监督管理局纳入优先审评品种名单,并于9月12日获得新药上市申请受理。据悉目前,禾元生物年产100万支奥福民的“黑灯工厂”已启动投产并获得《药品生产许可证》;年产120吨(1200万支)奥福民的“灯塔工厂”正在建设,预计2026年建成并投产。02 国内重组人血白蛋白在研情况白蛋白是人血浆中最丰富的蛋白质,占血浆总蛋白含量的50-60%,其最重要的生理功能包括维持渗透压、保持毛细血管通透性、稳定内皮细胞、转运和代谢多种化合物等,临床上主要用于补充体液和治疗低蛋白血症等。据安信证券数据披露,国内白蛋白的理论年总需求量约为1500-1800吨,但2021年全国批签发量为658吨,市场渗透率约为37%-44%。从全球层面来看,Allied Market Research数据预估,2020年全球人血清白蛋白治疗药物市场规模达到48.14亿美元,2030年预计达到89.56亿美元,复合年均增长率达到6.4%。全球人血白蛋白用量仍有发展空间,且亚太地区的复合年增长率预计会高于全球平均水平达到7.7%,但是供需矛盾一直是我国人血清白蛋白市场长期面临的问题。由于人血白蛋白需求量属于血液制品中最大的产品,仅靠国内产能生产无法满足患者用量,人血白蛋白也是唯一一个允许进口的血液制品,且主要依赖进口产品。而重组人血白蛋白(rHSA)是人血白蛋白的替代产品,研发目的之一是为了补足人血白蛋白市场空缺,期望可以达到国产人血白蛋白自给自足的状态。关于rHSA,国内外开展相关研究已数十年,但目前仍未出现在全球大范围内上市的成熟产品。至于国内,目前尚无重组人血白蛋白产品上市,现已知进展最快,有望在国内上市的产品就是上述提到的禾元生物和通化安睿特的重组人血白蛋白产品。其他国内在研企业还包括:深圳普罗吉医药:重组人血清白蛋白注射液处于Ⅲ期临床阶段,进行中但尚未开始招募,适应症为肝硬化腹水。健通生物:重组人血清白蛋白注射液处于Ia期临床,目标入组人数48人,目前已入组12人,适应症为肝硬化腹水患者低白蛋白血症。华北制药:辅料级重组人血白蛋白已获得国家食品药品监督管理局药品生产许可证。据华北制药举办的2024年半年报集体业绩说明会显示,基因重组人血白蛋白作为药用辅料用途已进入临床试验阶段,基因重组人血白蛋白药用注射剂正在进行临床前研究。 参考资料:[1]通化安睿特、禾元生物官微.[2]划时代——安睿特重组人白蛋白注射液Ⅲ期临床试验完成揭盲. 通化安睿特. 2025-04-16. [3]全球首发丨重组人血清白蛋白注射液(奥福民)III期临床达到研究终点. 禾元生物. 2024-11-30. [4]央视点名!这家生物制造,成功实现“稻米造血”,预计2025年量产!. 智药局. 2025-04-07. [5]一文带你了解重组白蛋白. 云岭血液制品. 2024-06-05. [6]全球唯一!救命药将面临骨折价. 药通社. 2024-04-19.识别微信二维码,可添加药时空小编请注明:姓名+研究方向!
临床3期临床成功上市批准加速审批
2024-12-16
Post author:alarpharm Post published:2024 年 12 月 16 日 Post category:最新消息
1.事實發生日:113/12/14 2.研發新藥名稱或代號:ALA-3000 注射劑 3.用途:難治型憂鬱症(Treatment-resistant depression, TRD) 4.預計進行之所有研發階段:一期臨床試驗/二期臨床試驗/三期臨床試驗/ 新藥查驗登記審核 5.目前進行中之研發階段(請說明目前之研發階段係屬提出申請/通過核准/不通過核准 ,若未通過者,請說明公司所面臨之風險及因應措施;另請說明未來經營方向及已投 入累積研發費用): (1)提出申請/通過核准/不通過核准/各期人體試驗(含期中分析)結果/發生其他 影響新藥研發之重大事件:本公司於今日接獲美國FDA通知核准ALA-3000注射劑 於難治型憂鬱症患者之臨床一期試驗申請。此臨床試驗為ALA-3000首次人體試 驗,用以評估ALA-3000在TRD患者的安全性、耐受性、藥物動力學、及初步的 療效探索,預計於美國招募約44位患者。 (2)未通過目的事業主管機關許可、各期人體臨床試驗(含期中分析)結果未達統計 上顯著意義或發生其他影響新藥研發之重大事件者,公司所面臨之風險及因應 措施:不適用。 (3)已通過目的事業主管機關許可、各期人體臨床試驗(含期中分析)結果達統計上 著意義或發生其他影響新藥研發之重大事件者,未來經營方向:不適用。 (4)已投入之累積研發費用:因涉及未來國際授權談判資訊,為避免影響授權金額 且為保障投資人權益,暫不揭露。 6.將再進行之下一階段研發(請說明預計完成時間及預計應負擔之義務): (1)預計完成時間:預計於2025年底前完成本試驗,實際執行時間依據收案狀況 而定。 (2)預計應負擔之義務:不適用。 7.市場現況: 難治型憂鬱症是重度憂鬱症的一種,被定義為當至少兩種不同的第一線抗憂鬱藥 物仍不足以控制憂鬱發作症狀的病況。根據國際期刊Psychiatric Quarterly指出 ,憂鬱症患者中有近三分之一的患者屬難治性憂鬱症。根據2021 National Survey on Drug Use and Health (NSDUH)的統計資料顯示,美國約有2,100萬成人曾經歷 過至少一次重度憂鬱症;在該年中約有1,450萬成人因重度憂鬱症而造成嚴重傷害。 市調機構Allied Market Research之調查資料指出,2020年全球抗憂鬱症藥物市場 規模為156.51億美元,預計到2030年將達210億美元,年複合增長率為3%。 8.其他應敘明事項(若事件發生或決議之主體係屬公開發行以上公司, 本則重大訊息同時符合證券交易法施行細則第7條第8款所定 對股東權益或證券價格有重大影響之事項):無。 9.新藥開發時程長、投入經費高且未保證一定能成功,此等可能使投資面臨風險,投 資人應審慎判斷謹慎投資。:
临床结果临床2期
2024-09-27
欢迎关注凯莱英药闻
近期,诺和诺德在基因编辑领域进一步加码;首先是在2024年6月,公司以最高4000万美元收购2seventy bio的A 型血友病项目及体内基因编辑技术的权益,后者可用于开发治疗自免疾病的自体或同种异体细胞疗法。其次,公司于近日达成两笔交易:分别是与Korro Bio达成高达5.3亿美元的药物合作,旨在开发两种此前无法成药靶点的RNA编辑候选药物,这些靶点适用于肥胖、糖尿病和心血管等疾病;以及与NanoVation达成一项多年研发合作伙伴关系,主要开发两个项目,未来有望扩展至多达五个针对心脏代谢性和罕见病的额外靶点,合作总金额高达6亿美元。
一
基因编辑及重要组成
基因编辑是指用可编辑的核酸酶识别基因组特定位点并介导DNA 双链断裂,随后诱发内源性DNA 修复机制,从而实现对DNA 序列的定点修饰的技术,包括靶向敲除或插入基因。基因编辑本质上是一种技术手段,由于多种疾病的发生与基因的错误表达直接相关,因此该技术有望实现疾病治疗。
完成基因编辑过程,无论是基因的敲除或插入,均需要:1)识别需要编辑的序列与点位,2)序列的剪切,3)剪切后的修复三个部分。工具、修复机制与载体构成了基因编辑的三大要素。
(一)基因编辑工具
目前,基因编辑工具手段经历了ZFNs 技术、TALENs 技术、CRISPR技术的更新迭代发展;完成序列的识别与剪切所依靠的基因编辑工具是各个企业研发重点,识别效率与剪切准确率是决定最终治疗效果的关键。
ZFNs(锌指核酸酶)技术是第一个普遍使用的基因编辑技术。ZFN 由锌指结构单元与Fok I 核酸酶活性区结合而成,锌指结构识别编辑位点,Fok I 核酸酶进行剪切。锌指结构由大约30 个氨基酸组成,同时与锌离子结合,因为结构与手指类似,因此称为锌指。由于针对不同的DNA序列,需要设计不同的锌指结构,较为费时费力。同时,由于识别位点较短,因此存在一定的脱靶现象,所以并未大规模应用于基因编辑治疗。
TALEN 技术与ZFNs 技术类似,DNA识别序列变为转录激活因子样效应因子(TALEs),核酸酶仍为Fok
I 核酸酶。与ZFNs 技术相比,具有特异性较高,识别精确的特点,但针对不同序列均需要重新开发TALE 结构,因此耗时费力,同时可能引起机体的免疫反应。
CRISPR 技术来源于细菌抵御外来噬菌体入侵的机制,细菌通过识别噬菌体的DNA 结构进而完成剪切。CRISPR 的识别DNA 序列由sgRNA完成,该结构由crRNA 和tracrRNA 两部分组成,切割由Cas9 蛋白完成。由于该机制中识别位点的特异性由RNA 决定而并非蛋白质,因此操作更加简单便宜,且可以剪切多个位点,是目前最便捷、应用最广泛的基因编辑工具。
(二)基因编辑载体
将工具递送至细胞内需要载体,较为成熟的载体有病毒递送载体、脂质纳米颗粒递送(LNP)、病毒样颗粒载体(VLP), 不同编辑策略需要选择合适的载体。
(三)治疗模式
此外,根据具体治疗的模式,基因编辑还可分为体内基因治疗与体外基因治疗。
体内基因编辑对技术精准度要求更高,可以做基因的敲除与插入;通过LNP等载体将基因编辑工具递送至患者体内特定组织与器官,进入细胞完成治疗过程;进度较为领先的代表公司为Intellia Therapeutics。
体外基因编辑强调对特定碱基或碱基序列的精确改写,其过程是将功能细胞在体外进行编辑,然后输回患者体内,完成治疗;目前全球首个获批的CRISPR 公司的Exa-cel,需要经过细胞提取,体外编辑,细胞回输,完成整个治疗过程。
二
近一年发生的交易
据不完全统计,2024年截至目前,在基因编辑疗法共发生10笔交易;已披露金额的主要是诺和诺德的3项交易以及国内企业信立泰与尧唐生物的发生交易,累计超13亿美元。
2024年8月,国内企业信立泰药业与尧唐生物就一款靶向PCSK9的碱基编辑药物YOLT-101签订协议。信立泰获得YOLT-101在中国大陆区域的独家许可权益,包括但不限于研发、注册、生产、商业化销售等;尧唐生物有望获批最高总交易金额近10.35亿元。YOLT-101尚处于临床前研究阶段,拟开发用于家族性高胆固醇血症(FH)等;已有的临床前数据显示,在非人灵长类动物模型中,YOLT-101实现了一次给药长达近两年的低密度胆固醇(LDL-C)大幅度降低。
三
管线及市场分析
据不完全统计,目前全球在研的基因编辑疗法约300余种;其中仅CRISPR/Cas9基因编辑疗法Casgevy获批。
处于临床以上阶段的基因编辑疗法
根据Allied Market Research以及Global Market Research统计,2021年至2022年,全球基因编辑行业市场规模由48.11亿美元增长至54.12亿美元,2022年同比增速为12.49%。根据Statista统计,2023年至2030年,全球基因编辑行业市场规模将以22.3%的复合增速进行增长,2023年市场规模约为66.19亿美元,到2030年,预计达到360.61亿美元。
四
重点公司及药物介绍
(一)CRISPR Therapeutics
CRISPR 是一家领先的基因编辑公司,专注于使用其专有的CRISPR/Cas9平台开发用于在治疗严重疾病的变革性基因药物。CRISPR在广泛的疾病领域建立了一系列治疗计划,其管线涉及血红蛋白病,肿瘤学,再生医学和罕见病。
公司第一代体外基因编辑产品的设计主要利用基因编辑技术增强了产品的安全性,避免不必要的免疫排斥。第二代体外基因编辑产品设计在第一代基础上的改进主要是为了增强工程细胞对肿瘤细胞的杀伤力。对不同基因的编辑,给细胞治疗带来了更大的设计空间,更多的改进方向。
CRISPR Therapeutics 降低排异反应的策略
重点药物介绍:Casgevy
Casgevy(通用名exagamglogene autotemcel,简称exa-cel)是CRISPR Therapeutics 与Vertex 联合研发的体外基因编辑疗法,开发适应症为Beta 地中海贫血和镰状细胞性贫血。通过体外基因编辑,研发团队对CD34+ HSPC 细胞中红细胞(erythroid)特异的BCL11A 基因的加强子(enhancer)进行抑制,以此得到改造后的细胞并给患者进行回输。BCL11A蛋白是一种调控因子,通常在出生后抑制胎儿血红蛋白的产生,因此通过BCL11A基因的编辑减少其蛋白产物的合成,可以促进造血干细胞产生更多携带胎儿血红蛋白的红细胞,以逐步取代成人的镰状红细胞。
Exa-cel 针对TDT和SCD 分别开展了临床研究,结果显示:在针对TDT 适应症的研究中,受试者接受exa-cel 治疗后,42/44 患者已停止输注红细胞。其中持续时间最长的已有36.2 个月没有进行输注;2 位还未停止输注的患者,所需输注的红细胞量已明显减少,分别下降了75%、89%。在针对SCD 适应症的研究中,接受exa-cel 治疗后,所有31 个患者均未出现血管闭塞危象VOC,其中持续时间最长的已有32.3 个月未出现VOC。
此外,Exa-cel 治疗后,含HbF 的细胞比例明显提升,TDT 组由基线的约10%提升至90%-100%区间,SCD组由基线的约20%提升至90%-100%。且提升后比例稳定维持在高位区间,TDT 组最长观察期已达36 个月,SCD组30 个月。
在安全性上,与白消安清髓术和自体造血干细胞移植(HSCT)相似。许多不良事件可能与配套的化疗(白消安)相关。
据生物医药行业媒体Endpoints News报道,Casgevy在美国的定价为220万美元(约合人民币1576万元)。
(二)Intellia Therapeutics
Intellia是基因编辑领域的明星企业,基于CRISPR/Cas9技术,针对Ex Vivo(离体)与In Vivo(在体)基因治疗建立了针对罕见遗传疾病、感染性疾病、免疫肿瘤学、血液疾病、自身免疫性疾病等领域的多样化在研管线,并取得优异的临床表现。
公司研发管线
公司通过敲除治疗细胞的TCR 来避免移植物排斥宿主病(GvHD);通过基因敲入引入CAR 或TCR来增强细胞对肿瘤的杀伤;敲除II 型HLA 基因来避免CD-4 介导的排异反应;敲除HLA-A 来避免CD-8 介导的排异反应;调整HLA-B 和HLA-C 来避免NK 细胞介导的排异反应。
Intellia 降低排异反应的策略
重点药物介绍:
1、NTLA-2001
NTLA-2001是全球进展最快的体内CRISPR基因编辑疗法,用于治疗甲状腺素转运蛋白(ATTR)淀粉样变性。此前 Intellia 已与再生元达成协议,Intellia将主导NTLA-2001的开发和商业化,再生元承担25%的成本,以换取相同的利润份额。目前,FDA已批准其体内CRISPR基因编辑疗法NTLA-2001开展关键3期临床试验,预计今年年底启动。
根据Intellia Therapeutiics 最新更新的临床结果,接受NTLA-2001治疗的所有转甲状腺素蛋白淀粉样变性患者的血清TTR 均降低≥90%,且这些获益一直持续到接受输注治疗后最长12 个月进行的最后一次访视。而接受25 mg 与75mg 剂量NTLA-2002 治疗的病患,平均激肽水平下降幅度为67%与95%,且分别持续了48 与32 周,显示出较好的治疗效果。
2、NTLA-2002
NTLA-2002通过抑制KLKB1基因表达,控制缓激肽的水平,降低血管扩张造成的水肿的风险,被开发用于治疗遗传性血管水肿(HAE)。目前,该药物已开展了2 项临床I期研究,分别针对由ATTR引起的多发性神经疾病和心肌疾病。
根据一项在患有遗传性血管性水肿的成年人开展的1 期剂量递增试验显示,在所有剂量水平下,最常见的不良事件是输液相关反应和疲劳。在施用NTLA-2002 后未观察到剂量限制性毒性作用、严重不良事件、3 级或更高不良事件。在基线和最新评估之间观察到总血浆激肽释放酶蛋白水平的剂量依赖性降低,25 mg组的平均百分比变化为 -67%,50 mg组为 -84%,75 mg组为 -95%。从基线到第 1 至第 16 周(主要观察期),25 mg组每月血管性水肿发作次数的平均百分比变化为 -91%,50 mg组为 -97%,75 mg组为 -80%。在所有患者中,从基线到最新评估,每月血管性水肿发作次数的平均百分比变化为-95%。
(三)Editas Medicine
EditasMedicine 成立于2013 年,这是一家基于 CRISPR/Cas9 专利技术开发针对遗传疾病和癌症基因编辑治疗的公司,也是第一家上市的 CRISPR 基因编辑公司。公司致力于将CRISPR/Cas9和CRISPR/Cas12a基因组编辑系统的能力和潜力转化,为治疗世界各地严重疾病患者的药物。
Editas
Medicine 研发管线
重点药物介绍:EDIT-301
EDIT-301(renizgamglogene
autogedtemcel,又称reni-cel)是Editas
Medicine 正在开发中的治疗镰状细胞病和β-地中海贫血的CRISPR基因编辑疗法,对患者来源的CD34+造血干细胞和祖细胞进行基因编辑,编辑位点是HBG基因的启动子区域,这是因为HBG基因的启动子区域是转录抑制因子BCL11A的结合位点,靶向改为点就能抑制BCL11A,从而重新激活γ-珠蛋白的表达,改善患者的红细胞成熟。该疗法使用的是CRISPR-Cas12a基因编辑系统,即Cas12a和gRNA组成的RNP(RNA和蛋白质复合物),通过电穿孔技术将其转染到细胞中,以避免使用病毒载体带来的随机插入风险。
根据2024年5月公司在2024 EHA公布的数据,在一项 I/II 期、多中心、开放标签、单臂研究,旨在评估 reni-cel 对严重 SCD 患者的安全性、耐受性和疗效。截至 2024 年 2 月,17 名可评估患者在输注reni-cel 后的平均(标准差,SD)时间为 6.2(5.8)个月;其中两名患者的随访时间超过 1 年。中性粒细胞和血小板植入分别在平均(SD)22.2(3.8)天和 25.2(6.2)天后实现。没有患者报告输注 reni-cel 后出现 VOE,而入组前 2 年内平均(SD)每年出现 5.2(3.0)次严重 VOE(n=17)。输注 reni-cel 后,平均(SD)Hb水平迅速升高,并从第 5 个月起维持在 ≥14.4(2.5)g/dL(n=8)。到第 4 个月(n=7),HbF 的平均(SD)百分比为 48.0% (4.0),且在最后一次随访中维持在 >40%。F 细胞和MCH-F/F 细胞的百分比早期增加,并从第 5 个月(n=8)起维持在 >98% F 细胞,从第 3 个月(n=7)起维持在 >15 pg/F 细胞,这代表了具有临床意义的目标。所有患者的溶血关键标志物均得到改善或正常化。reni-cel 的安全性与使用白消安的清髓性预处理一致。未报告与 reni-cel 相关的不良事件。
(四)Beam:基于碱基编辑的基因治疗公司
Beam Therapeutics成立于2018年,基于单碱基编辑技术,通过重写基因组中的单个碱基,在目标DNA序列上产生精确的可预测和有效的基因修饰。公司目前正在研发的基因治疗递送策略包括:电穿孔 、 非病毒载体 (主要是LNP)和 病毒载体 (主要是AAV)三种。公司计划使用电穿孔技术进行体外血细胞和免疫细胞的递送 (例如治疗地中海贫血症等血液性疾病) ,使用LNP进行肝脏和其他器官的体内递送,使用AAV进行眼睛和中枢神经系统的递送。
Beam Therapeutics 研发管线
重点药物介绍:BEAM-101
目前公司进度最快的产品为BEAM-101,一种体外碱基编辑疗法,用于治疗镰状细胞病。该一次性疗法包括自体 CD34+ 造血干细胞和祖细胞 (HSPC),这些细胞已在 HBG1/2 基因的启动子区域进行了碱基编辑,并通过造血干细胞移植程序进行给药。BEAM-101编辑旨在抑制转录抑制因子 BCL11A 与启动子结合,而不会破坏 BCL11A 表达,从而增加非镰状和抗镰状胎儿血红蛋白 (HbF) 的产生,从而模仿遗传性胎儿血红蛋白持续性中自然发生的变异的影响。
(五)Verve Therapeutics
Verve Therapeutics 致力于开发一种治疗心血管疾病(CVD)的方法,将治疗从慢性管理转变为单疗程基因编辑药物。该公司最初的两个项目分别针对调节血脂水平的基因PCSK9和ANGPTL3。
Verve Therapeutics的研发管线
重点药物介绍:Verve-101
公司主要候选产品Verve-101是全球第一个进入临床的、在人体内部进行碱基编辑的疗法,利用基于CRISPR系统改造的单碱基编辑器,改变患者细胞中PCSK9基因的一个碱基,达到让PCSK9失活的效果。PCSK9是降低LDL-C的热门靶点,抑制它活性的功效已经得到了多款FDA批准疗法的验证。
2024 年 4 月,公司公布了其在13 名患者中的给药数据,显示对于 0.45 mg/kg 组的前五名随访至少 28 天的受试者,平均 LDL-C 降低范围从 21% 到 73%,平均为 46%(截至数据截止日期 2024 年3 月)。在 0.45 mg/kg 或 0.6 mg/kg 组中随访时间最长的两名患者中,LDL-C 降低持续长达 270 天,且随访仍在进行中。
(六)Prime Medicine
Prime Medicine于2022年10月在纳斯达克上市,主要围绕一种全新的精准基因编辑工具——先导编辑(Prime Editor,PE),该技术无需依赖DNA模板便可有效实现所有12种单碱基的自由转换,而且还能有效实现多碱基的精准插入与删除。2023年5月,刘如谦团队在Nature
Biotechnology期刊发表论文,对先导编辑进行了改进和升级——双先导编辑(TwinPE),能够实现大片段DNA的删除、替换、整合和倒位,为治疗复杂人类遗传疾病或大型基因突变所致的人类遗传疾病提供了有效工具。
重点药物介绍:PM359
PM359 是公司在血液学和免疫学重点领域推出的首个候选产品,针对的是慢性肉芽肿病 (CGD) 的 p47phox 变体,这是一种在儿童时期出现的严重且危及生命的疾病。PM359 包含使用 Prime Editors 进行体外修饰的自体造血干细胞 (HSC),旨在纠正大量含有致病突变的细胞。目前,PM359 已获得美国食品药品管理局 (FDA) 授予的罕见儿科药物称号和孤儿药称号。
根据2024年5月公布的数据,临床前研究显示:(1)纠正了 75% 以上 CGD 患者 CD34+ 细胞中的 CGD 致病突变;(2)在小鼠模型中,≧80% 的Prime Edited CGD 患者 CD34+ 细胞中的CGD 致病突变得到纠正;(3)在植入Prime Edited CGD 患者 CD34+ 细胞的小鼠中,骨髓中性粒细胞中的 NADPH 氧化酶活性得到恢复;(4)与未编辑的 CGD 患者细胞相比,Prime Edited CGD 患者细胞中干扰素调节基因表达降低,与健康供体细胞相似,表明 Prime Editing 将细胞恢复到健康状态;(5)在植入的 Prime Edited CGD 患者 CD34+ 细胞中未检测到非预期或脱靶编辑。
(七)国内公司发展情况
国内基因编辑企业有望加速发展;截至2023年12月,我国已经有50余家公司涉足基因编辑技术,但其中依然以初创公司为主,管线研发也多在临床早期,如尧唐生物、博雅辑因、邦耀生物、瑞风生物、辉大基因、本导基因等。
1、尧唐生物
尧唐生物成立于2021年,由华东师范大学生命科学学院研究员吴宇轩等人于2021年创立,是一家专注于结合mRNA体内递送技术和基因编辑技术,开发新一代mRNA药物和基因编辑药物的高科技生物技术公司。尧唐生物通过对CRISPR、碱基编辑和其他新一代基因编辑工具的持续开发和优化,对新一代mRNA生产平台和脂质纳米载体组装工艺的创新型改进,致力于开发针对遗传性疾病和心血管疾病的体内基因编辑药物。
2、博雅辑因
博雅辑因是一家专注基因编辑技术转化的、处于临床阶段的生物医药企业,由北京大学生命科学学院教授魏文胜创立。公司基于在基因编辑技术、高通量基因组编辑筛选和生物信息等方面的科研和技术实力,推进国际领先的体外和体内基因编辑疗法开发。公司已经建立了包括体外疗法造血干细胞平台、体外疗法通用型CAR-T平台、体内疗法RNA碱基编辑平台在内的多个治疗平台。
公司体外基因编辑疗法 ET-01是中国首个获国家药监局批准开展临床试验的基因编辑疗法产品和造血干细胞产品,用于治疗输血依赖型β地中海贫血。
3、邦耀生物
邦耀生物致力于成为新商业文明时代全球领先的细胞基因药企,由华东师范大学生命科学学院前院长、生命医学研究所所长刘明耀于2013年创立,依托自主研发中心及与高校共建的“上海基因编辑与细胞治疗研究中心”,目前已产生100多项专利成果,有5个项目在8所知名医院开展研究者发起的临床试验,3个项目已获批IND,正式进入注册临床试验阶段,还有多个项目进入IND申报阶段。
4、本导基因
本导基因是一家基因治疗创新药物研发企业,由上海交通大学系统生物医学研究院教授蔡宇伽等人于2018年创立,致力于为眼科、神经系统、造血系统、病毒感染以及肿瘤等多领域的难治性疾病开发具有全球意义的创新药物。本导基因拥有国际领先VLP mRNA递送平台(BDmRNA)和下一代慢病毒载体平台(BDlenti)。围绕着核心递送技术平台,本导基因布局了多条first-in-class的产品管线,开展了多项first-in-human临床研究,与国际著名药企开展了重大神经系统疾病新型疗法的合作研发。
5、其它
还有多家国内企业布局新型CRISPER基因编辑疗法,例如正在进行碱基编辑疗法开发的正序生物、贝斯生物、新芽基因,进行表观遗传基因组编辑疗法开发的益杰立科等。
参考资料
1、各公司官网
2、招商证券、浙商证券、中银国际
3、澎湃新闻、前瞻网、生物世界、医药魔方
感谢关注、转发,转载授权、加行业交流群,请加管理员微信号“hxsjjf1618”。
“在看”点一下
基因疗法并购细胞疗法
100 项与 Allied Scientific Products 相关的药物交易
登录后查看更多信息
100 项与 Allied Scientific Products 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年05月21日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
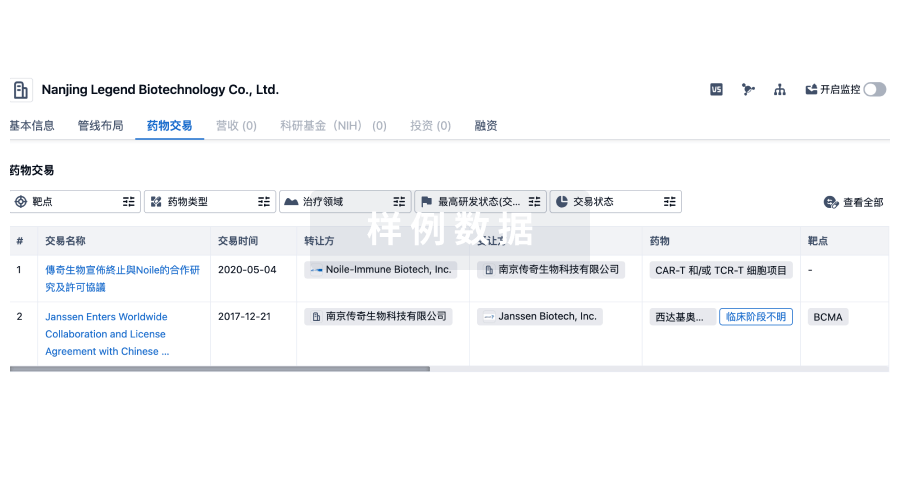
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
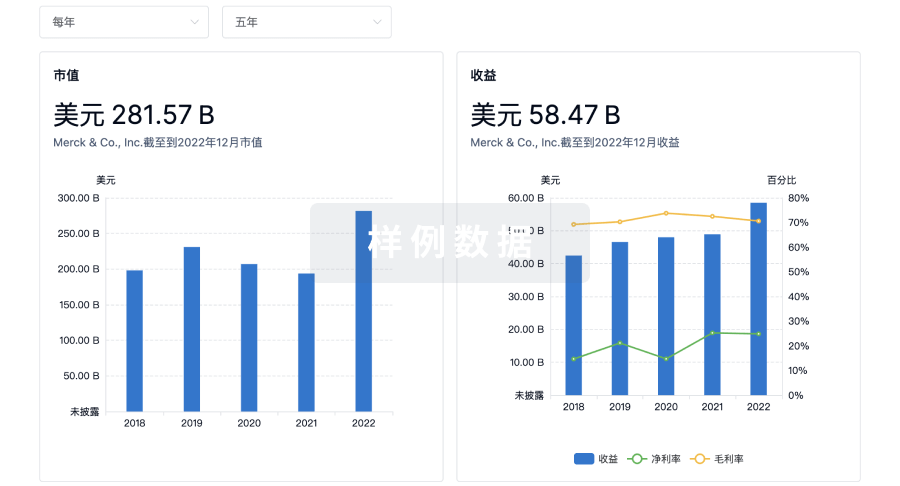
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用