预约演示
更新于:2026-02-19
Beijing Daxiang Biotech Co., Ltd.
更新于:2026-02-19
概览
关联
100 项与 北京大橡科技有限公司 相关的临床结果
登录后查看更多信息
0 项与 北京大橡科技有限公司 相关的专利(医药)
登录后查看更多信息
93
项与 北京大橡科技有限公司 相关的新闻(医药)2026-02-17
·微信
点击蓝字 关注我们
癌症治疗的核心困境在于肿瘤异质性带来的个体疗效差异,传统治疗难以兼顾疗效与安全性。类器官(由患者肿瘤细胞体外培养而成的微型肿瘤模型,能精准模拟原发肿瘤的异质性和微环境特征)与纳米医学的结合,为解决这一问题提供了新路径。类器官可在体外精准复刻患者肿瘤的生物学特征,纳米医学则能实现药物的靶向递送与可控释放。
最新发表在《Materials Today Bio》(IF=10.2)的综述研究指出,二者的协同应用正在重塑癌症治疗模式:类器官为纳米药物的筛选提供了贴近临床的体外模型,解决了传统动物模型与人体肿瘤差异大的问题;纳米技术则突破了类器官筛选结果向临床转化的递送瓶颈,实现了从体外筛选到体内治疗的闭环。本文将系统解析这一技术组合的核心突破、应用场景及发展方向,展现癌症治疗向精准化、个体化迈进的核心路径。
01
研究亮点
Research Highlights
1. 技术协同创新:类器官精准复刻肿瘤异质性与微环境,纳米医学实现靶向递送与可控释放,二者构建 “体外筛选-体内治疗” 闭环,使纳米药物临床转化成功率提升,大幅降低研发失败风险。
2. 个性化治疗落地:依托多组学数据与AI算法,结合类器官高通量筛选,定制专属纳米药物方案,肿瘤抑制率高,且中性粒细胞减少等毒副作用显著降低,突破传统治疗 “一刀切” 局限。
3. 多场景突破瓶颈:不仅加速纳米药物研发,还破解实体瘤递送难题,赋能化疗+免疫等联合治疗,为血脑屏障跨越、多药耐药逆转等提供新路径,支撑癌症精准治疗多维度突破。
02
研究内容
Research Content
1. 癌症纳米医学的主要进展
1.1癌症纳米医学概述
癌症纳米医学是融合纳米技术、生物材料学与制药工程的精准治疗平台,通过1-100nm纳米载体的独特理化性质,实现药物高负载、靶向递送及脱靶效应降低(图 1)。其核心优势在于高比表面积带来的高载药量、靶向递送减少脱靶效应,以及支持化疗、基因治疗、光热治疗等多模式联合应用。
其载体类型丰富,包括脂质体、聚合物胶束、无机纳米颗粒、外泌体等,可通过被动富集、主动靶向或刺激响应机制,将化疗药物、核酸、免疫调节剂等递送至肿瘤组织深处。给药方式也呈现多样化,包括鼻黏膜沉积、可溶解微针经皮递送、肿瘤内储库注射、口服纳米晶体制剂等,每种方式均利用独特的解剖学途径最大化局部药物暴露,同时减少全身脱靶效应。
图1 癌症治疗中选择性纳米医学药物递送系统概述及其优势。左上:各类纳米载体(脂质体、聚合物胶束、无机纳米颗粒等)的结构示意图;右下:纳米医学相较于传统治疗的核心优势,包括增强溶解性与生物相容性、靶向递送、多药联合治疗、利用肿瘤微环境触发激活、克服生物屏障、规避耐药性等。
目前全球已有至少15种纳米药物获批,80余种候选药物正在开展超过200项临床试验(表1和表2)。获批药物中,脂质体类占比最高,如1995年获批的多柔比星脂质体(Doxil®)、2005年获批的白蛋白结合型紫杉醇(Abraxane®)、2015年获批的伊立替康脂质体(Onivyde®)等,主要用于乳腺癌、卵巢癌、胰腺癌等治疗。临床管线中,53%为I/II期研究,以肿瘤适应症为主,涵盖被动/主动靶向及刺激响应等多种策略,但也面临高淘汰率挑战,如HER2靶向脂质体多柔比星(MM-302)因体内靶向优势不足未能通过III期试验。
表1 获批的癌症纳米药物汇总(精选)
表 2 处于不同临床试验阶段
用于肿瘤治疗的代表性纳米药物
1.2 纳米药物的智能靶向策略
纳米医学主要采用被动和主动靶向策略将药物递送至肿瘤(图2A)。被动靶向依赖增强渗透滞留效应(EPR效应),即纳米颗粒通过渗漏的新生血管渗出,并因淋巴引流功能障碍而滞留,使载药在肿瘤部位富集,同时避免损伤正常器官。EPR介导的富集程度主要取决于载体直径(10-200nm)、zeta电位(±10-30mV)和形状(如球形、棒状、盘状或蠕虫状),而非分子识别。该尺寸范围可避免肾脏清除,减少组织驻留巨噬细胞的摄取,同时允许纳米颗粒通过内皮间隙。
但EPR效应具有高度异质性,在不同肿瘤类型、分期和个体患者间差异显著,例如在软组织肉瘤中比在致密的胰腺癌中更明显。为应对这一问题,可通过EPR成像辅助剂(如超顺磁性氧化铁MRI或放射性标记脂质体)进行患者分层,识别潜在应答者;或联合血管启动剂(如放疗),以患者特异性方式暂时改善血管通透性,提升递送效率。
与这些适应性策略并行,主动靶向为克服被动EPR的局限性提供了更直接的方法。通过在纳米颗粒表面修饰配体、抗体、肽、适体或小分子,使其与恶性细胞或肿瘤相关血管上过表达的受体结合,主动靶向旨在实现不依赖血管通透性的特异性。结合后,受体介导的内吞作用将载药直接递送至细胞质,提高细胞内药物浓度并规避外排泵。为覆盖更多肿瘤亚群,研究人员正在开发多价构建体,携带两种或多种识别互补受体的不同配体,从而扩大靶向范围并提高结合亲和力。此类策略的实施需要对每个肿瘤进行详细的分子谱分析,以确保配体-受体相容性并最小化脱靶效应。
除配体导向富集外,纳米医学目前已利用肿瘤微环境(TME)本身作为精准释放的生物触发信号。癌症纳米医学智能递送的刺激信号主要分为两类(图2B):内源性刺激响应型纳米颗粒在肿瘤微环境的弱酸性胞外pH(6.5-6.9)、毫摩尔级谷胱甘肽浓度、肿瘤富集蛋白酶或升高的过氧化氢(H2O2)等信号触发下,通过特殊设计的连接键(腙键、二硫键、肽键)发生结构坍塌或化学断裂,确保载药仅在恶性细胞内释放;外源性刺激响应型纳米颗粒则由近红外光、超声、交变磁场、电离辐射或电脉冲触发,可实现正交的、操作者可控的激活。光热脂质体、磁加热氧化铁核心、超声断裂囊泡和X射线激活闪烁体等平台,可按需释放药物或产生细胞毒性热/活性氧,不受肿瘤异质性影响。通过整合内源性和外源性刺激,下一代纳米医学正发展为“逻辑门控”递送系统,需要双重或序贯信号激活,从而在最大限度提高肿瘤特异性的同时保护健康组织完整性。
图2 纳米药物智能靶向示意图。(A)纳米药物的靶向机制示意图:左图为通过增强渗透滞留效应(EPR 效应)实现被动靶向,右图为通过配体或抗体修饰纳米颗粒实现主动靶向;(B)智能癌症纳米药物递送的刺激信号分类,包括外源性刺激和内源性刺激。
此外,生物启发靶向是另一种重要的药物递送方式,包括利用肿瘤微生物组酶解激活的微生物组调控纳米医学,以及依托外泌体天然靶向性的生物衍生载体,兼具生物相容性与靶向精准性。
表3 纳米药物智能靶向策略总结表
1.3 癌症纳米医学与人工智能(AI)
实体瘤的患者间、肿瘤内及转移灶间异质性,导致传统“一刀切”纳米药物难以实现统一治疗窗口。个性化癌症纳米医学以患者特异性多组学数据(基因组学、转录组学、蛋白质组学等8类)为基础,结合AI算法,将肿瘤异质性转化为纳米载体的定量设计规则,包括最优尺寸、表面化学、靶向配体及刺激响应模式。
AI与机器学习算法(如随机森林、卷积神经网络)可整合多组学数据,预测纳米颗粒生物分布与治疗响应;生成对抗网络则能辅助设计新型纳米载体。而患者来源类器官(PDOs)作为核心验证模型,能精准复刻原发肿瘤的异质性、微环境特征及药物响应,可用于纳米药物的高通量筛选与疗效验证,显著提升临床转化成功率,成为连接多组学设计与临床应用的关键桥梁。
2. 类器官:纳米药物的“临床前试金石”
传统纳米药物研发常陷入“临床前有效,临床无效”的困境——动物模型难以复刻人类肿瘤的异质性,导致大量在小鼠身上表现优异的纳米载体,进入人体后因无法适应复杂的TME而失效。类器官的出现,恰好填补了这一转化鸿沟。
2.1 精准复刻肿瘤特征,突破传统模型局限
类器官由患者肿瘤组织体外培养而成,能完整保留原发肿瘤的细胞亚群、血管结构雏形和微环境特征(如酸性pH、高谷胱甘肽浓度等),其基因表达谱与原发肿瘤高度相似。研究数据显示,基于类器官筛选的纳米药物,临床转化成功率比传统动物模型提升3-5倍(图 3)。
这种精准模拟能力让纳米药物筛选更贴近临床实际。例如在乳腺癌研究中,高还原型 / 氧化型谷胱甘肽比值的肿瘤类器官,对二硫键连接的纳米缀合物响应更显著;而组织蛋白酶B高表达的类器官,则对聚L-谷氨酸(PGA)连接的多柔比星敏感性更高,这一发现直接指导了后续纳米药物的个性化选择。
图3 类器官用于纳米药物筛选的核心流程与结果a:基底膜提取物(BME)包埋的乳腺癌类器官制备流程;b:不同亚型乳腺癌类器官的尺寸分布散点图;c:线性聚L-谷氨酸(PGA)纳米载体与类器官的共定位成像(绿色为纳米载体,红色为溶酶体标记物,蓝色为细胞核);d:共定位信号峰值重叠的荧光强度定量分析;e:不同纳米载体在类器官中的时间依赖性摄取曲线;f:组织蛋白酶B在类器官中的表达水平检测;g:不同亚型乳腺癌类器官的形态学特征;h:游离多柔比星(Dox)与PGA偶联多柔比星(PGA-hyd-Dox)对不同类器官的细胞活力抑制曲线。
2.2 支持高通量筛选,助力机制解析
类器官可实现规模化培养,配合自动化检测技术,能同时对数十种纳米载体进行药效评估。研究团队通过类器官模型发现,纳米颗粒的尺寸、表面电荷和刺激响应机制,需与肿瘤的具体特征匹配才能发挥最佳效果:针对缺氧型肿瘤类器官,pH/ROS双响应纳米载体的药物释放效率比单一响应载体提升40%;而对间质致密的胰腺癌类器官,表面修饰透明质酸的纳米颗粒穿透深度可增加2.3倍。
更重要的是,类器官能揭示纳米药物的作用机制。通过对类器官的单细胞测序分析,研究者发现纳米药物不仅能直接杀伤肿瘤细胞,还能重塑肿瘤微环境——例如MnO₂基纳米探针可通过耗竭谷胱甘肽、放大活性氧,在类器官中诱导“爆发性铁死亡”,同时激活抗肿瘤免疫信号,这为联合治疗方案设计提供了直接依据。
3. 纳米医学+类器官:个性化治疗的协同机制
类器官的体外筛选结果需通过高效递送才能落地临床,纳米技术恰好提供了解决方案。二者的协同,让癌症治疗真正实现 “量体裁衣”—— 根据患者类器官的特征,定制专属纳米药物,再通过纳米载体的靶向递送,实现疗效最大化、毒性最小化。
3.1 类器官指导纳米药物精准定制
每个患者的肿瘤类器官都具有独特的分子特征,这些特征成为纳米药物设计的“导航坐标”。相关研究中,研究者通过分析患者类器官的pH值、酶表达水平和受体丰度,设计了三种个性化纳米药物方案(图4):
针对高表达叶酸受体的卵巢癌类器官,设计叶酸修饰的pH响应脂质体,药物在肿瘤部位的富集量比未修饰载体提高3.7倍;
对高表达MMP-2酶的乳腺癌类器官,采用酶响应型聚合物胶束,实现药物在肿瘤部位的特异性释放,减少脱靶毒性;
对多药耐药(MDR)类器官,构建“化疗药物+siRNA”共递送纳米载体,既通过siRNA沉默P-糖蛋白基因,又实现化疗药物的靶向递送,逆转耐药率达68%。
临床前数据显示,基于类器官定制的纳米药物,在患者来源异种移植(PDX)模型中的肿瘤抑制率达85%以上,而传统纳米药物的抑制率仅为42%-58%,且定制化方案的毒副作用显著降低——中性粒细胞减少发生率从31%降至7%,心脏毒性从12%降至2%。
图4 卵巢癌个性化纳米医学治疗蓝图。a:癌症治疗从“一刀切”到个性化的发展轨迹示意图;b:靶向纳米载体设计;c:四种卵巢癌患者样本的肿瘤体积响应曲线;d:卵巢癌个性化纳米治疗方案流程图。
3.2 纳米技术突破临床转化障碍
类器官虽能精准模拟肿瘤,但如何将基于类器官筛选的药物高效递送至体内肿瘤部位,仍需纳米技术的助力。研究中,纳米载体通过三种方式突破体内屏障:
首先是主动靶向,利用类器官鉴定出的高表达受体(如HER2、CD44),在纳米颗粒表面修饰对应的配体或抗体,实现“受体-配体”介导的精准结合。例如针对CD44高表达的三阴性乳腺癌类器官,透明质酸修饰的纳米颗粒能通过CD44受体介导的内吞作用,使肿瘤部位药物浓度提升5.2倍(图5)。
图5 酶响应与双重靶向纳米药物系统。其次是刺激响应释放,根据类器官检测的TME特征(如酸性 pH、高谷胱甘肽),设计智能纳米载体,进入肿瘤部位后才触发药物释放。例如针对pH 6.2的胃癌类器官,腙键连接的纳米载体在肿瘤部位的药物释放率达78%,而在正常组织(pH 7.4)中释放率仅为12%。
最后是联合治疗递送,将化疗药物、免疫调节剂等多种payload共封装于纳米载体,实现“一载体多功效”。例如负载RSL3(铁死亡诱导剂)和diABZi(STING激动剂)的Bi₂Se₃纳米颗粒,在类器官指导下,既能通过RSL3诱导肿瘤细胞铁死亡,又能通过diABZi激活全身免疫应答,在三阴性乳腺癌模型中实现原发肿瘤抑制和转移灶清除(图6)。
图9 辐射响应与电场响应纳米医学系统。a:DP-HBN/RA纳米药物的作用机制,X射线触发活性氧爆发和GPX4阻断,放大铁死亡并激活免疫系统;b:14天小鼠肺转移成像,DP-HBN/RA+X射线组转移灶最少;c:4T1肿瘤生长曲线,DP-HBN/RA+X射线组实现持续消退;d:肺组织H&E染色,DP-HBN/RA+X射线组转移灶稀少。
3.3 闭环治疗体系:从类器官到临床应用
研究者构建了“类器官筛选-纳米药物定制-临床治疗-疗效监测”的闭环体系(图7):首先通过患者肿瘤组织构建类器官,筛选最优纳米药物方案;然后根据类器官的药效数据,确定临床给药剂量和频率;治疗过程中,通过重复活检更新类器官模型,动态调整纳米药物设计,确保治疗效果持续优化。
图7 个性化癌症纳米医学的多组学驱动流程从样本收集(组织、尿液、粪便、血液等体液活检)、多组学分析(基因组学、转录组学、蛋白质组学等8类组学)、核心检测技术(靶向测序、WES、WGS、单细胞RNA测序等)、数据整合分析(机器学习/AI算法+纳米药物设计+类器官验证)、临床前验证(患者来源类器官、患者来源异种移植模型)到规模化生产(GLP毒理学研究、GMP生产)和临床应用(I-III期临床试验)的完整流程。
在卵巢癌的初步临床探索中,这一闭环体系使患者的客观缓解率(ORR)从传统治疗的23%提升至57%,无进展生存期(PFS)从5.3个月延长至11.7个月,且未出现严重不良反应,初步验证了该体系的临床可行性。
4. 类器官+纳米医学:多场景突破癌症治疗瓶颈
除了个性化治疗,类器官与纳米医学的融合还在基础研究、药物研发和联合治疗等多个场景发挥重要支撑作用,为解决癌症治疗的核心难题提供了新方案。
4.1 加速纳米药物研发,降低失败风险
传统纳米药物研发周期长、成本高,约90%的候选药物因临床前与临床差异而失败。类器官的介入,让研发流程实现“提质增效”——在药物发现阶段,利用类器官快速筛选有效纳米载体,淘汰无效方案;在临床前评估阶段,通过类器官预测药物的临床疗效和毒性,提高临床试验成功率。
研究显示,引入类器官筛选的纳米药物研发,周期缩短了30%-40%,研发成本降低了25%-35%。例如针对结直肠癌的超声响应纳米药物,通过类器官筛选,快速确定了最优纳米颗粒尺寸(100nm)和超声参数(1MHz),后续临床试验的成功率比传统研发模式提高了2倍。
4.2 破解实体瘤治疗难题,突破生物屏障
实体瘤的致密间质和高interstitial压力,是纳米药物递送的主要障碍。类器官模型帮助研究者找到针对性解决方案:通过分析胰腺癌类器官的间质特征,设计了“基质重塑+药物递送”双功能纳米载体——表面修饰透明质酸酶的脂质体,既能降解肿瘤间质的透明质酸,降低间质压力,又能递送化疗药物,使药物穿透深度从肿瘤表面的20μm提升至100μm以上(图8)。
图8 纳米药物递送与肿瘤组学的关联分析示意图。
此外,针对血脑屏障这一药物递送的关键屏障,研究者通过脑胶质瘤类器官筛选,发现修饰转铁蛋白受体(TfR)的 pH 响应纳米颗粒,能高效跨越血脑屏障,在脑肿瘤部位的药物浓度比未修饰载体提高6.8倍,且能在酸性TME中触发药物释放,显著抑制胶质瘤生长。
4.3 赋能联合治疗,提升疗效深度
癌症的复杂性决定了单一治疗难以根治,类器官与纳米医学的融合,让联合治疗更精准、更高效。研究中,研究者通过类器官筛选,确定了多种协同效果显著的联合方案:
纳米化疗+免疫治疗:负载化疗药物的纳米颗粒诱导肿瘤细胞免疫原性细胞死亡(ICD),同时递送PD-L1抑制剂,在黑色素瘤类器官模型中,CD8+T细胞浸润量增加3.5倍,肿瘤抑制率达92.9%;
纳米光热治疗+化疗:金纳米壳介导的光热治疗,在类器官中被证实可破坏肿瘤血管,增强化疗药物渗透,联合治疗的疗效比单一治疗提升2.8倍;
纳米基因治疗+化疗:siRNA沉默KRAS G12D基因的纳米载体,与化疗药物共递送,在胰腺癌类器官模型中,肿瘤细胞凋亡率达76%,而单一治疗的凋亡率仅为32%-45%。
这些联合方案的协同机制,均通过类器官模型得以验证,为临床联合治疗策略的制定提供了直接依据。
03
局限性与展望
Limitations and Prospects
尽管类器官与纳米医学的融合已取得显著突破,但要实现广泛临床应用,仍需克服三大核心挑战:首先是类器官模型的完整性问题。目前的类器官难以完全复刻体内肿瘤的血管系统和免疫微环境,可能导致纳米药物的体内外效果存在偏差。未来需通过共培养免疫细胞、内皮细胞等,构建更接近体内真实情况的“类肿瘤生态系统”,提升体外筛选结果的预测准确性。其次是纳米药物的规模化生产难题。个性化纳米药物的小批量生产面临成本高、批间差异大等问题,限制了其临床普及。解决方案包括开发模块化纳米载体平台,通过更换靶向配体和活性成分,快速适配不同患者的需求;同时建立GMP级别的小型化生产体系,降低生产成本,提升可及性。最后是临床转化的监管与伦理障碍。个性化纳米药物的审批缺乏统一标准,且患者肿瘤组织的获取和类器官构建涉及伦理规范。这需要监管机构建立专门的审批通道,同时制定类器官研究的伦理指南,明确样本收集、使用和数据共享的规范,确保技术合规应用。
展望未来,随着AI技术的介入,这一领域将迎来更大突破:通过AI分析类器官的多组学数据,自动设计最优纳米药物方案;结合可穿戴设备实时监测患者病情,动态调整纳米药物的剂量和递送方式,最终实现“实时个性化治疗”。类器官与纳米医学的融合,不仅改变了癌症治疗的技术路径,更重塑了治疗理念——从药物适配患者转向患者定制药物。随着技术的持续迭代,癌症治疗将逐步进入精准化、个体化的新时代,为患者带来更高疗效、更低毒性的治疗选择。
类器官与纳米医学的协同创新,正在破除癌症治疗的核心困境,推动肿瘤学从标准化治疗向精准治疗转型。这一技术组合的核心价值,在于以患者的肿瘤特征为核心,通过类器官实现精准筛选,借助纳米技术实现高效递送,构建了从体外模型到体内治疗的完整体系。
扫描二维码可下载原文
大橡科技肿瘤类器官药物评价
大橡科技依托肿瘤类器官样本库,打造精准药物评价解决方案。高度还原肿瘤微环境与异质性,突破传统模型局限,可高效支持小分子、抗体药物、ADC、CGT等候选药物的敏感性检测与耐药研究,助力药企加速筛选进程,降低研发成本与试错风险,同时为临床个体化用药赋能,推动肿瘤药物研发与精准医疗落地。
(详情请点击下方链接)
客户文章解读系列|IF40.8的前列腺癌治疗突破研究!eRNA驱动铁死亡,联合疗法攻克去势抵抗性难题
客户文章解读系列|Cancer Research从预测到增效:肿瘤类器官免疫模型助力免疫亚型分类系统创新
客户文章解读|类器官模型验证TFE3重排肾细胞癌治疗新机制
珍芯严®全场景解决方案
你有思路,我来想!你出方案,我来做!
以专业眼光洞察数据,让决策更加清晰!
珍芯严®面向药企药物早期研发与高校、医院的基础科研,我们倾力打造类器官芯片全场景解决方案。凭借创新的类器官芯片技术,打破传统细胞与动物研究瓶颈,高效产出优质数据,从实验室到临床,我们与您并肩作战!
客服电话/微信:18610798010
扫描二维码可联系我们
小红书
设计: Summer、Rebecca|封面: Rebecca
求点赞
求分享
求喜欢
基因疗法
2026-02-15
·微信
点击蓝字 关注我们
在肿瘤基础医学与转化研究中,胰腺导管腺癌(PDAC)犹如一团难以驱散的阴霾,以其高度的侵袭性、复杂的微环境和极高的致死率,长期占据着“癌中之王”的宝座。尽管针对其核心驱动基因KRAS的靶向疗法已初现曙光,但肿瘤细胞通过复杂的代谢重编程机制产生的适应性耐药,依然是横亘在临床治愈道路上的鸿沟。在这一背景下,能够高度还原患者肿瘤异质性、微环境特征及药物反应表型的类器官(Organoids)模型,正逐渐成为解码肿瘤代谢、筛选精准治疗策略的关键。类器官技术不仅在体外重现了肿瘤的三维结构,更关键地保留了原代肿瘤的分子分型与代谢特征,为验证新型治疗策略的广谱有效性提供了无可替代的实验平台。
2026年1月16日,来自德国弗莱堡大学医学中心(Medical Center - University of Freiburg)的研究团队,在国际顶尖学术期刊《Signal Transduction and Targeted Therapy》(STTT,影响因子>30)上发表了一项题为“Vertical RAS pathway inhibition in pancreatic cancer drives therapeutically exploitable mitochondrial alterations”的重磅研究。该研究并未止步于传统的细胞系筛选,而是将目光投向了更能代表临床真实情况的患者来源类器官(Patient-Derived Organoids, PDOs)与基因工程小鼠模型(KPC),旨在从代谢重塑的深层机制中寻找突破RAS抑制耐药的新路径。
研究团队聚焦于RAS信号通路的垂直抑制策略——即联合抑制上游的磷酸酶SHP2与下游的激酶MEK1/2。这种联合策略虽然在既往研究中显示出延缓耐药的潜力,但其诱发的深层代谢后果却鲜为人知。研究团队通过多组学分析、超微结构观察及功能实验,惊人地发现:RAS通路的垂直抑制并非单纯地“饿死”肿瘤,而是通过诱导剧烈的线粒体重塑(Mitochondrial Remodeling),迫使肿瘤细胞进入一种代谢极度活跃但氧化应激防御极其脆弱的状态。这种状态为诱导铁死亡(Ferroptosis)——一种铁依赖性的脂质过氧化驱动的细胞死亡形式——打开了治疗窗口。更令人振奋的是,研究团队利用类器官模型证实,这种代谢脆弱性并不受限于PDAC复杂的分子亚型(如基底样型或经典型),从而为临床上异质性极强的胰腺癌患者提供了一种极具潜力的通用型联合治疗策略。
01
研究亮点
Research Highlights
1. 首次系统性阐明了RAS通路垂直抑制会导致PDAC细胞发生深刻的线粒体结构与功能重塑,表现为线粒体质量异常增加、备用呼吸能力代偿性提升及ROS水平激增,从而产生对脂质过氧化清除途径(特别是GPX4)的致命依赖。
2. 类器官验证了治疗策略的“跨亚型”广谱性:利用涵盖不同转录组亚型(Basal-like vs. Classical)的患者来源类器官(PDOs),强有力地证实了诱导铁死亡是克服RAS抑制耐药的通用策略。
3. 确立SHP2/MEK/铁死亡三联疗法的体内疗效:在原本难以治疗的KPC自发成瘤小鼠模型中,确立了“SHP2抑制剂 + MEK抑制剂 + 天然铁死亡诱导剂(Withaferin A)”的三联疗法,该组合显著抑制了肿瘤进展,为临床难治性胰腺癌提供了极具转化潜力的联合用药方案。
02
研究内容
Research Content
1. RAS通路垂直抑制:打破耐药的必经之路与代谢代价
胰腺导管腺癌(PDAC)的基因组图谱中,超过90%的病例携带KRAS基因的激活突变。KRAS蛋白作为细胞内的分子开关,控制着细胞增殖、分化、存活以及至关重要的代谢重编程。长期以来,直接靶向KRAS曾被认为是不可成药的挑战,尽管近年来KRAS G12C等特定突变体的抑制剂取得了突破,但针对PDAC中更常见的G12D、G12V突变的广谱抑制仍面临巨大困难,且单一靶点的抑制极易通过反馈回路的激活导致耐药。
在此背景下,研究者们采取了垂直抑制(Vertical Inhibition)的策略。Src同源2结构域磷酸酶2(SHP2,由PTPN11基因编码)是连接受体酪氨酸激酶(RTK)与RAS-MAPK通路的关键接头蛋白,对于KRAS完全激活至关重要。研究团队推测,同时阻断SHP2(上游)和MEK1/2(下游效应器),可以双重夹击RAS信号流,防止因MEK抑制导致的RTK反馈性激活,从而实现更持久的通路抑制。
为了全面评估这一策略,研究者构建了一个庞大的体外筛选平台,包含了人类PDAC细胞系(MIA PaCa II, PANC-1, YAPC)以及来源于KPC转基因小鼠的鼠源性细胞系。这些细胞系经过严格的转录组学分类,覆盖了PDAC两大核心分子亚型:基底样型(Basal-like/Squamous)和经典型(Classical/Progenitor)。基底样型通常预后更差,对化疗更耐药;而经典型则保留了更多上皮特征。
药物敏感性筛选结果显示,虽然单药SHP2抑制剂(SHP099)对细胞增殖的影响微乎其微,单药MEK抑制剂(Trametinib)在不同细胞系中表现出异质性反应,但SHP2与MEK的联合抑制在所有测试细胞系中均产生了显著的协同致死效应,成功克服了部分细胞系的内源性耐药。然而,这种强效的信号抑制并非没有代价,它迫使肿瘤细胞为了生存而进行剧烈的代谢重组,这正是该研究的核心切入点(图1)。
2. 代谢靶点高通量筛选
为了捕捉RAS通路垂直抑制诱导的代谢脆弱性,研究团队设计了一套精密的代谢抑制剂筛选流程。他们在维持SHP2/MEK抑制的背景下,分别针对糖酵解、磷酸戊糖途径(PPP)、自噬、脂肪酸合成及氧化、线粒体呼吸等关键代谢通路进行了干扰测试。结果揭示了一幅复杂的代谢重塑图景:
糖酵解(Glycolysis):使用2-DG抑制糖酵解在基底样型细胞中显示出更高的敏感性,这与基底样型肿瘤更依赖糖酵解供能的既往认知相符。
自噬(Autophagy):自噬抑制剂氯喹(Chloroquine)在绝大多数细胞系中均增强了RAS通路抑制的杀伤效果,且这种增敏效应在联合抑制下比单药MEK抑制更为显著。这提示肿瘤细胞在信号通路受阻时,急需通过自噬回收胞内物质以维持生存。
然而,最引人注目的发现来自于线粒体(Mitochondria)。筛选数据显示,当RAS通路被垂直阻断时,肿瘤细胞对线粒体复合物I抑制剂(Rotenone)和ATP合酶抑制剂(Oligomycin)的敏感性在所有亚型中均显著增加。通过Seahorse能量代谢分析仪进行的实时检测进一步解开了谜团:SHP2/MEK联合抑制导致肿瘤细胞的备用呼吸能力(Spare Respiratory Capacity)显著提升。
这似乎是一个悖论:被抑制的肿瘤细胞为何反而增强了线粒体功能?研究指出,这实际上是一种代偿性的应激反应。为了应对生长信号的阻断和糖酵解的受限,肿瘤细胞被迫挖掘线粒体的潜能,通过增加线粒体质量(Mitochondrial Mass)和重组电子传递链来维持能量稳态。流式细胞术证实了联合治疗后线粒体质量的普遍增加,同时也伴随着一个危险的副产物——活性氧(ROS)水平的激增。这种线粒体反扑虽然暂时延续了细胞的生命,却也将细胞推向了氧化应激(图1)。
图1. 药理学SHP2和/或MEK抑制重编程PDAC细胞代谢。(a) PDAC细胞系的转录组分类:通过整合形态学特征、蛋白表达水平及转录组基因集富集分析(GSEA),将人类(MIA PaCa II, PANC-1, YAPC)和鼠源(KPC系列)PDAC细胞系定位于从“基底样(Basal-like)”到“经典型(Classical)”的分子谱系上。这一分类为后续评估代谢抑制剂的亚型特异性奠定了基础。(b) 细胞增殖实验:展示了SHP099(SHP2抑制剂,15 μM)、Trametinib(MEK抑制剂,10 nM)单药及联合用药对不同细胞系的抑制效果。数据表明,联合用药普遍克服了单药耐药性,在所有测试细胞系中均显示出最强的生长抑制。(c) 代谢抑制剂筛选流程:左图为实验设计示意图,展示了如何在MAPK通路抑制(SHP2i/MEKi)背景下,叠加使用不同浓度的代谢抑制剂。右图为代表性的结晶紫染色板,展示了在长时间培养(6-14天)后,不同组合对细胞密度的影响。(d-g) 代谢抑制剂敏感性热图:(d) 糖酵解抑制(2-DG)在基底样型细胞中效果更显著,而联合RAS抑制可增敏经典型细胞。(e) 自噬抑制(Chloroquine)在联合RAS抑制背景下,对几乎所有细胞系均显示出强烈的增敏效应。(f) 脂肪酸合成抑制(C75)抑制所有细胞,但未见与RAS抑制的明显协同;脂肪酸氧化抑制(Etomoxir)主要影响基底样型。(g) 线粒体呼吸抑制(Oligomycin, Rotenone)及PGC-1α抑制在联合RAS抑制下,对所有亚型细胞均显示出显著的增敏效应,提示线粒体是关键的代谢依赖点。(h) 线粒体质量评估:流式细胞术(MitoTracker Deep Red)结果显示,SHP099/Trametinib联合治疗导致无论是基底样型还是经典型细胞,其线粒体质量(Mitochondrial mass)均显著增加。(i) 活性氧(ROS)水平检测:流式细胞术检测显示,联合治疗后细胞内总ROS水平趋于升高,提示氧化应激增加。(j) 线粒体功能分析(Seahorse):氧耗率(OCR)分析显示,联合治疗显著提高了细胞的备用呼吸能力(Spare respiratory capacity),表明细胞为了应对治疗压力,增强了线粒体的代谢潜能。
3. 多组学图谱绘制:氨基酸代谢流与转录组的全面重编程
为了将观察到的表型变化与分子机制联系起来,研究团队利用非靶向代谢组学、靶向蛋白质组学和转录组学技术,绘制了RAS通路垂直抑制下的全景分子图谱。
代谢组学(Metabolomics)分析聚焦于细胞内外的代谢物交换。主成分分析(PCA)显示,联合治疗72小时后,细胞外液的代谢指纹发生了剧烈偏移。最显著的变化在于氨基酸代谢:细胞外液中大量氨基酸(如丝氨酸、缬氨酸)累积,提示细胞对氨基酸的摄取能力下降。然而,与氧化还原平衡密切相关的代谢物却呈现出截然不同的趋势。细胞内的谷胱甘肽(GSH)、S-腺苷同型半胱氨酸(SAH)以及甲硫氨酸亚砜(MSO)水平显著下降,而这些代谢物在细胞外液中却有所增加。这种胞内谷胱甘肽等抗氧化代谢物的减少,结合线粒体ROS水平的升高,共同表明细胞处于氧化应激状态,不得不消耗大量的谷胱甘肽来中和线粒体过量产生的ROS。
转录组学(Transcriptomics)数据的基因集富集分析(GSEA)进一步印证了这一结论。联合治疗显著下调了与DNA/RNA复制及蛋白质合成相关的基因集,这与细胞增殖停滞的表型一致。相反,与脂质代谢(Lipid Metabolism)、类固醇代谢以及线粒体氧化磷酸化(OXPHOS)相关的基因集则表现出显著的代偿性上调。这种转录层面的重编程清晰地描绘了一个处于饥饿但亢奋状态的细胞:一方面试图通过增强线粒体呼吸和脂质利用来获取能量,另一方面却因ROS的累积而面临脂质过氧化的风险。
蛋白质组学(Proteomics)则捕捉到了线粒体动态相关蛋白的改变,以及铁死亡关键调节因子GPX4和FSP1的表达波动。特别是GPX4的表达在部分细胞系中受到抑制,这为后续锁定铁死亡作为治疗靶点提供了直接线索(图2)。
图2. 多组学揭示RAS通路抑制下的细胞代谢重塑。(a) 胞外代谢物主成分分析(PCA):展示了MIA PaCa II等细胞系在经过DMSO、SHP099、Trametinib或联合治疗72小时后,细胞外液代谢组分的巨大差异。联合治疗组与其他组明显分离。(b) PCA贡献因子分析:展示了对PCA分离贡献最大的代谢物类别,其中氨基酸(Amino acids)是最主要的贡献者,表明氨基酸摄取与消耗模式的改变是治疗响应的核心特征。(c) 胞内/胞外代谢物热图:对比了SHP2/MEK抑制下,特定代谢物在胞内和胞外的丰度变化。关键发现是谷胱甘肽(GSH)、S-腺苷同型半胱氨酸(SAH)、甲硫氨酸亚砜(MSO)等与氧化还原及甲硫氨酸循环相关的代谢物,在胞内显著减少,而在胞外显著增加,提示细胞抗氧化能力的耗竭和代谢物的泄漏。(d) 转录组GSEA分析:KEGG通路富集气泡图。颜色代表标准化富集分数(NES)。结果显示,联合治疗显著下调了DNA/RNA复制、核苷酸代谢及氨基酸代谢通路(与增殖受阻一致),同时显著上调了脂质代谢、类固醇代谢、自噬(Autophagy)及线粒体氧化磷酸化(OXPHOS)相关基因集,揭示了转录层面的代谢重编程方向。
4. 深入体内微环境:TIF分析与单细胞测序揭示普遍脆弱性
为了在更复杂的体内微环境中验证上述发现,研究团队使用了KPC转基因小鼠模型。这是一种能够自发形成胰腺导管腺癌的小鼠,其肿瘤具有完整的免疫微环境和致密的基质,是公认的最难治疗但也最接近人类疾病的模型。
研究团队采用了一种极具创新性的技术——肿瘤组织间液(Tumor Interstitial Fluid, TIF)分析。TIF是肿瘤细胞、基质细胞与免疫细胞进行物质交换的直接场所,其代谢成分比外周血更能准确反映肿瘤局部的代谢状态。通过液相色谱-串联质谱(LC-MS/MS)分析KPC小鼠肿瘤的TIF,研究发现联合治疗组的TIF中,与线粒体呼吸和ROS防御相关的氨基硫醇(Aminothiols)水平发生了显著变化。特别是半胱氨酸、甲硫氨酸及其代谢产物SAM/SAH的水平异常,直接指向了甲硫氨酸循环和转硫通路的代偿性激活。这表明,在体内缺氧、缺营养的恶劣环境下,RAS通路抑制同样引发了肿瘤细胞对氧化还原平衡维持机制的极度依赖(图3)。
单细胞RNA测序(scRNA-seq)技术进一步解析了肿瘤内部的异质性。PDAC肿瘤通常由上皮样(Epithelial)和间质样(Mesenchymal)等多种亚群组成,它们对药物的反应往往不同。然而,scRNA-seq分析显示,无论是上皮样还是间质样的肿瘤细胞亚群,在联合治疗后均表现出ROS相关通路基因及氧化磷酸化基因的显著富集。这一结果至关重要,它排除了代谢重塑仅发生于特定细胞亚群的可能性,证实了线粒体重塑和氧化应激是RAS通路垂直抑制诱导的普遍性细胞事件,不论细胞处于何种分化状态(图4)。电镜下的超微结构观察为这一结论提供了证据。在体内治疗后的肿瘤组织中,可以清晰地观察到癌细胞内线粒体体积增大、基质致密、线粒体脊(Cristae)密度增加。这种形态学的改变正是线粒体功能亢进和代谢压力增加的典型特征。
图3. 体内肿瘤微环境与线粒体超微结构改变。(a) KPC小鼠肿瘤动力学:展示了用于提取肿瘤组织间液(TIF)的KPC小鼠肿瘤在治疗期间的体积变化曲线,确认了所选样本对治疗有响应。(b) TIF代谢物分析:柱状图展示了与Vehicle组相比,联合治疗组TIF中各代谢物的变化。关键发现包括氨基酸(如缬氨酸、亮氨酸)及神经递质水平的升高,以及半胱氨酸、甲硫氨酸、SAM、SAH等含硫代谢物的显著改变,佐证了体内的氧化应激反应。(d) 线粒体直径量化:基于电镜图像的统计数据。每个点代表一个线粒体。结果显示,无论是短期还是长期联合治疗,肿瘤细胞内的线粒体直径均显著大于对照组。(e) 线粒体超微结构(电镜图):代表性的透射电镜(TEM)图像。展示了联合治疗组肿瘤细胞内线粒体体积增大,且线粒体脊(Cristae)排列更加致密,呈现出典型的高代谢活性形态。
图4. 单细胞测序揭示肿瘤细胞异质性中的普遍代谢特征。(a) 单细胞亚群聚类(UMAP):展示了从KPC小鼠肿瘤中分离的数万个单细胞的聚类结果。重点展示了上皮样肿瘤细胞(Epithelial clusters, E1-E5)和间质样肿瘤细胞(Mesenchymal clusters, M1-M3)的分布。(c) 亚型评分:展示了不同细胞亚群在Collisson、Moffitt等经典PDAC分子分型体系下的评分,确认了E亚群对应经典型,M亚群对应基底样型。(d) 治疗组分布:展示了不同治疗组细胞在各亚群中的比例。长期Trametinib治疗导致间质样细胞比例增加,而联合治疗组则显示出不同的分布模式。(f) 亚群特异性代谢基因富集:热图展示了在联合治疗后,不同细胞亚群中代谢通路的富集情况。关键结果是:几乎所有的肿瘤细胞亚群(包括E和M亚群),均表现出ROS相关通路(ROS associated pathways)及氧化磷酸化(OXPHOS)基因的显著上调。这证明了线粒体重塑和氧化应激是跨越细胞表型异质性的普遍治疗反应。
5. 类器官(PDOs)验证:跨越分子亚型的铁死亡易感性
基于上述多维度的证据,研究团队推导出一个关键假设:RAS通路抑制导致线粒体过度活跃和ROS积累,必然使细胞高度依赖抗氧化系统来防止脂质过氧化(Lipid Peroxidation)。如果此时人为阻断关键的脂质过氧化清除酶——谷胱甘肽过氧化物酶4(GPX4),是否能引爆细胞内的ROS炸弹,诱导灾难性的铁死亡(Ferroptosis)?
为了验证这一假设并评估其临床转化潜力,研究团队利用了极其珍贵的资源——胰腺癌患者来源类器官(Patient-Derived Organoids, PDOs)。研究团队从手术切除的PDAC组织中分离肿瘤细胞,在基质胶(Matrigel)中进行3D培养,成功建立了多个PDO株系。通过转录组测序,这些类器官被严格划分为基底样型(Basal-like)、经典型(Classical)以及中间型(Intermediate)。这种分类至关重要,因为在临床上,基底样型患者通常预后极差,且对常规化疗不敏感,而经典型则相对较好。一个理想的治疗策略必须能够覆盖这两种截然不同的亚型。
实验结果的突破性发现:
脂质过氧化水平的本底升高:利用C11-BODIPY荧光探针检测发现,仅使用SHP2/MEK抑制剂处理类器官,即可观察到细胞膜脂质过氧化水平的显著升高。这证实了RAS通路抑制确实将细胞推向了铁死亡的边缘。
GPX4抑制剂的合成致死效应:当在SHP2/MEK抑制剂的基础上,联合使用GPX4特异性抑制剂ML210时,几乎所有的PDO株系均表现出爆发式的细胞死亡。这种杀伤效果远远超过了单药或双药治疗。
跨亚型的广谱有效性:最令人振奋的是,这种合成致死效应在不同亚型的类器官中均得到了验证。无论是通常难以治疗的基底样型类器官(如PDO-B100, B150),还是经典型类器官,均无法逃脱铁死亡的命运。这表明,诱导铁死亡是针对RAS通路抑制后代谢脆弱性的通用解法,不受PDAC分子分型的限制。
此外,研究还测试了直接靶向KRAS G12D的抑制剂(MRTX1133)和泛RAS抑制剂(RMC-7977)。结果显示,直接阻断RAS同样会诱发线粒体应激和脂质过氧化依赖,联合GPX4抑制剂同样能产生强协同效应。这进一步拓展了该策略的应用范围,意味着无论是垂直抑制还是直接抑制,只要阻断了RAS通路,铁死亡诱导剂都能成为其最佳拍档(图5)。
图5. RAS通路抑制诱导脂质过氧化依赖及类器官验证。(a) 脂质过氧化原位检测:左图为代表性的MDA(丙二醛)免疫组化染色图,右图为H-score量化分析。结果显示,联合治疗组肿瘤组织中MDA水平显著高于对照组,表明体内发生了严重的脂质过氧化。(b) 铁死亡敏感性筛选(C11-BODIPY):热图展示了在多种PDAC细胞系中,不同处理下的脂质过氧化水平。单纯RAS通路抑制(RMC-4550/Trametinib)即导致脂质过氧化水平升高(黄色/红色区域);当加入GPX4抑制剂ML210时,脂质过氧化水平呈爆发式增长(深红色),且该效应可被铁死亡抑制剂Ferrostatin-1逆转。(f) 类器官(PDO)转录组分型:热图展示了11个患者来源类器官株系的分子分型,明确标注了基底样型(Basal-like)、经典型(Classical)和中间型(Intermediate)。(h-i) 类器官增殖实验:(h) 柱状图展示了PDO在不同处理下的相对增殖率。(i) 散点图总结了GPX4抑制剂ML210在不同亚型类器官中带来的额外杀伤效应(Add-on effect)。结果表明,无论是SHP2/MEK联合抑制,还是泛RAS抑制剂(RMC-7977),叠加ML210后均能在所有亚型的类器官中产生显著的协同致死效应(平均增效10-44%),证实了该策略的广谱性。
6. 临床前转化:三联疗法在体内遏制肿瘤进展
从体外类器官走向体内实体瘤治疗,是转化医学的关键一步。然而,由于目前的GPX4小分子抑制剂(如ML210)在体内的药代动力学性质较差,不适合直接用于动物实验。因此,研究团队选择了一种天然产物——Withaferin A (WA)。WA是从茄科植物南非醉茄(Withania somnifera)中提取的甾体内酯。既往研究表明,WA不仅是一种天然的铁死亡诱导剂,还能通过抑制Hsp90等机制发挥抗癌作用,且具有较好的生物安全性。
在KPC小鼠模型中,研究团队设计了“SHP2抑制剂(SHP099) + MEK抑制剂(Trametinib) + Withaferin A”的三联给药方案。MRI长期监测显示,与单纯的双药联合(SHP2i + MEKi)相比,加入WA的三联疗法显著延缓了肿瘤体积的增长。对治疗后的肿瘤组织进行免疫组化分析,发现三联治疗组肿瘤中的丙二醛(MDA,脂质过氧化的终产物)水平显著升高,直接证实了铁死亡在体内的发生。生存分析显示,三联疗法延长了小鼠的无进展生存期。然而,由于KPC小鼠本身体质虚弱,大多数小鼠死于治疗相关的虚弱而非肿瘤负荷过大。
这一结果虽然提示了毒性管理的挑战,但更有力地证明了代谢联合治疗在抑制肿瘤生长方面的强大潜力。它表明,只要能开发出毒性更低、靶向性更强的铁死亡诱导剂,这一策略就有望转化为临床上的治愈性疗法(图6)。
图6. 三联疗法在体内模型的疗效评估。(a) 治疗方案示意图:展示了KPC自发成瘤小鼠的入组、MRI监测及给药流程。治疗组包括Vehicle、双药(SHP099+Trametinib)、单药Withaferin A以及三药联合(双药+Withaferin A)。(b-c) 肿瘤体积监测:展示了治疗开始前各组肿瘤的基线体积及治疗期间的体积变化。(d) 肿瘤生长曲线:左图:肿瘤重量/体重比。右图:基于MRI测量的肿瘤相对体积变化曲线。结果显示,三联疗法(红色曲线)相比双药疗法(蓝色曲线),进一步显著抑制了肿瘤体积的增长,曲线最为平缓。(f) 生存分析(Kaplan-Meier):展示了各组小鼠的生存曲线。尽管双药治疗已显著延长生存期,三联疗法在控制肿瘤进展方面表现最佳,但由于小鼠体质原因(治疗相关毒性),总生存期的进一步延长受到限制(HR分析显示三联疗法对比Vehicle有显著获益)。
03
未来展望
Future Outlook
这项发表于2026年的前沿研究,通过精细的机制解析和多模型验证,为胰腺癌治疗描绘了一幅全新的代谢蓝图。其核心价值在于发现RAS信号通路的抑制不仅仅是阻断了增殖信号,更是一次剧烈的细胞代谢重构事件,这种重构暴露了致命的代谢弱点。
该研究再次凸显了类器官在药物研发中的独特价值。与传统的2D细胞系相比,类器官保留了患者特异性的遗传背景和表观遗传特征。该研究中,正是利用了PDOs覆盖不同分子亚型的能力,才得以确证铁死亡策略的广谱性。在未来,对于每一位PDAC患者,利用其肿瘤样本构建类器官库,并进行RAS抑制剂与代谢调节剂(如铁死亡诱导剂)的联合筛选,将成为实现个体化代谢治疗的标准流程。类器官将不仅是科研工具,更是临床决策的“替身”。
虽然该研究使用了天然产物Withaferin A作为概念验证,但其药理性质仍非完美。这一发现将极大地激励制药工业界开发更专一、更稳定、毒性更低的GPX4抑制剂或新型铁死亡诱导剂(如FSP1抑制剂、CoQ10类似物等)。针对RAS通路抑制后的特定代谢状态(高ROS、高线粒体负荷)设计的智能递送系统,也将是未来的热点。
传统的抗癌思路往往局限于“阻断通路”,而该研究提出了诱导脆弱性并加以利用的新思路。即通过RAS抑制剂将癌细胞驱赶至一种高氧化应激状态,再利用铁死亡诱导剂给予致命一击。这或将成为解决KRAS突变肿瘤耐药问题的通用法则,不仅适用于胰腺癌,也可能推广至肺癌、结直肠癌等其他RAS驱动的肿瘤。
综上所述,这项研究不仅为RAS通路垂直抑制剂的临床应用提供了代谢层面的理论依据,更通过类器官模型确立了铁死亡作为胰腺癌联合治疗关键靶点的地位,为攻克这一“癌中之王”带来了新的曙光。
扫描二维码可下载原文
大橡科技肿瘤类器官药物评价
大橡科技依托肿瘤类器官样本库,打造精准药物评价解决方案。高度还原肿瘤微环境与异质性,突破传统模型局限,可高效支持小分子、抗体药物、ADC、CGT等候选药物的敏感性检测与耐药研究,助力药企加速筛选进程,降低研发成本与试错风险,同时为临床个体化用药赋能,推动肿瘤药物研发与精准医疗落地。
(详情请点击下方链接)
客户文章解读系列|IF40.8的前列腺癌治疗突破研究!eRNA驱动铁死亡,联合疗法攻克去势抵抗性难题
客户文章解读系列|Cancer Research从预测到增效:肿瘤类器官免疫模型助力免疫亚型分类系统创新
客户文章解读|类器官模型验证TFE3重排肾细胞癌治疗新机制
珍芯严®全场景解决方案
你有思路,我来想!你出方案,我来做!
以专业眼光洞察数据,让决策更加清晰!
珍芯严®面向药企药物早期研发与高校、医院的基础科研,我们倾力打造类器官芯片全场景解决方案。凭借创新的类器官芯片技术,打破传统细胞与动物研究瓶颈,高效产出优质数据,从实验室到临床,我们与您并肩作战!
客服电话/微信:18610798010
扫描二维码可联系我们
小红书
设计: Summer、Rebecca|封面: Rebecca
求点赞
求分享
求喜欢
临床研究
2026-02-13
·今日头条
> 2026年2月13日,国内类器官芯片领域的先行者北京大橡科技有限公司宣布完成逾亿元B轮融资。
这笔由北京市医药健康产业投资基金和北京经济技术开发区产业升级股权投资基金二期联合投资的资金,不仅为公司发展注入动力,更被视为类器官芯片技术从实验室走向产业化应用的关键转折点。
## 融资聚焦AI与自动化,瞄准四大战略方向
本轮融资资金将重点投向四个核心领域:**AI驱动的类器官芯片技术迭代**、类器官芯片生命科学仪器智造升级、智能化药物筛选与精准医疗平台搭建,以及数字孪生与虚拟实验技术攻关。
这标志着大橡科技的战略重心从单一技术验证,转向构建“**类器官芯片+AI+自动化**”的深度融合生态,旨在加速该技术在生物医药全产业链的规模化落地。
## 技术核心:全闭环商业化与三大业务品牌
大橡科技成立于**2018年**,是**国内首家实现类器官芯片“设计-建模-研发-生产-应用”全闭环商业化**的企业。其自主研发的 **IBAC®(集成仿生阵列芯片)平台**拥有完全自主知识产权,通过干细胞与微流控技术在体外精准复现人体组织器官的结构与功能。
基于该平台,公司构建了覆盖药物研发到临床治疗的三大业务品牌:
- **珍芯严®**:为靶点发现、药物评价和机制研究提供临床相关性强的体外模型。
- **安可芯®**:支持肿瘤精准治疗的用药决策及治疗方案优化。
- **橡芯®**:以**AI与自动化**为核心,推动类器官芯片设备的标准化与智能化升级。
## 产业化成果:高精度模型与临床前验证
经过数年发展,大橡科技已积累扎实的产业化基础。公司已推出**50余种**类器官培养试剂盒,构建**100余种**人源化病生理模型,并开发**6套**核心仪器设备。这些成果已直接支撑**4项创新药管线完成临床IND(新药临床试验)申报**,并在部分环节实现了**动物实验替代**。
在模型验证方面,大橡科技牵头制定了肾和肿瘤模型的验证方案,并参与肝、肠、心脏模型验证。揭盲结果显示,其**肠芯片、肾芯片、肝芯片的预测准确度分别达到100%、100%和90%**。这一数据为类器官芯片作为可靠药物评价工具提供了关键背书。
## 行业东风:监管认可与生态构建
类器官芯片技术的产业化正迎来全球政策东风。国际上,**美国FDA**相关政策已明确将基于类器官芯片等替代方法的非临床评价结果纳入快速审批通道;国内,**国家药监局药品审评中心(CDE)** 也已发布5项技术指导原则支持其应用。
> “类器官芯片技术已从单点科研验证阶段进入平台能力与应用场景并行推进的关键时期。”
与此同时,大橡科技积极构建产业生态,已与**国内外数百家知名药企、三甲医院及科研机构**建立合作,并发布了国内首个标准化类器官样本库 **OrganoidBOX**,致力于打造共建共享的应用生态圈。
## 资本视角:押注产业升级的关键“使能技术”
此次联合投资的双方均从产业高度肯定了类器官芯片的价值。**北京经济技术开发区产业升级股权投资基金**表示,类器官芯片是赋能生物医药产业升级的关键 **“使能技术”** ,投资大橡科技将强化区域在下一代生物技术竞争中的核心优势。
从科研工具到产业基础设施,大橡科技的此次融资超越了单一企业事件,折射出生物医药研发范式向更高效、更精准、更人性化方向演进的大趋势。
随着“类器官芯片+AI+自动化”技术的持续迭代与成本下降,其在新药发现、毒性评价及个性化医疗中的应用边界有望不断拓宽,最终影响全球药物研发的效率与格局。
100 项与 北京大橡科技有限公司 相关的药物交易
登录后查看更多信息
100 项与 北京大橡科技有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月10日管线快照
无数据报导
登录后保持更新
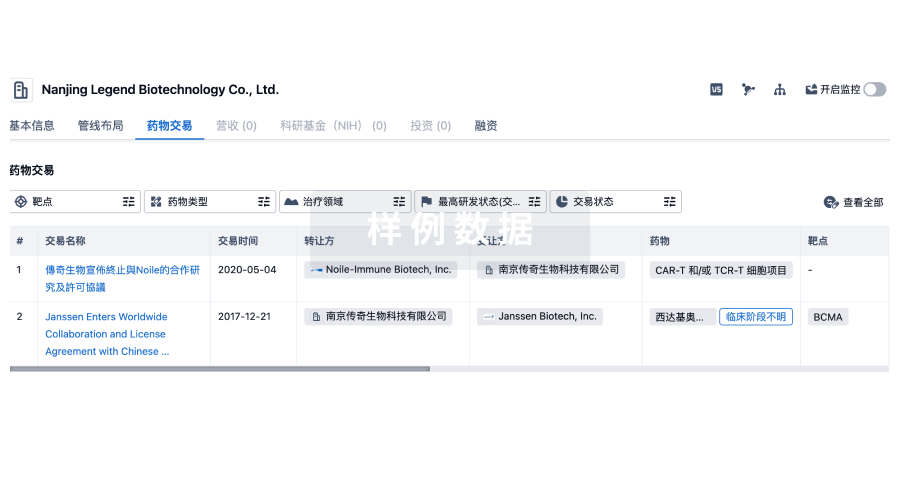
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
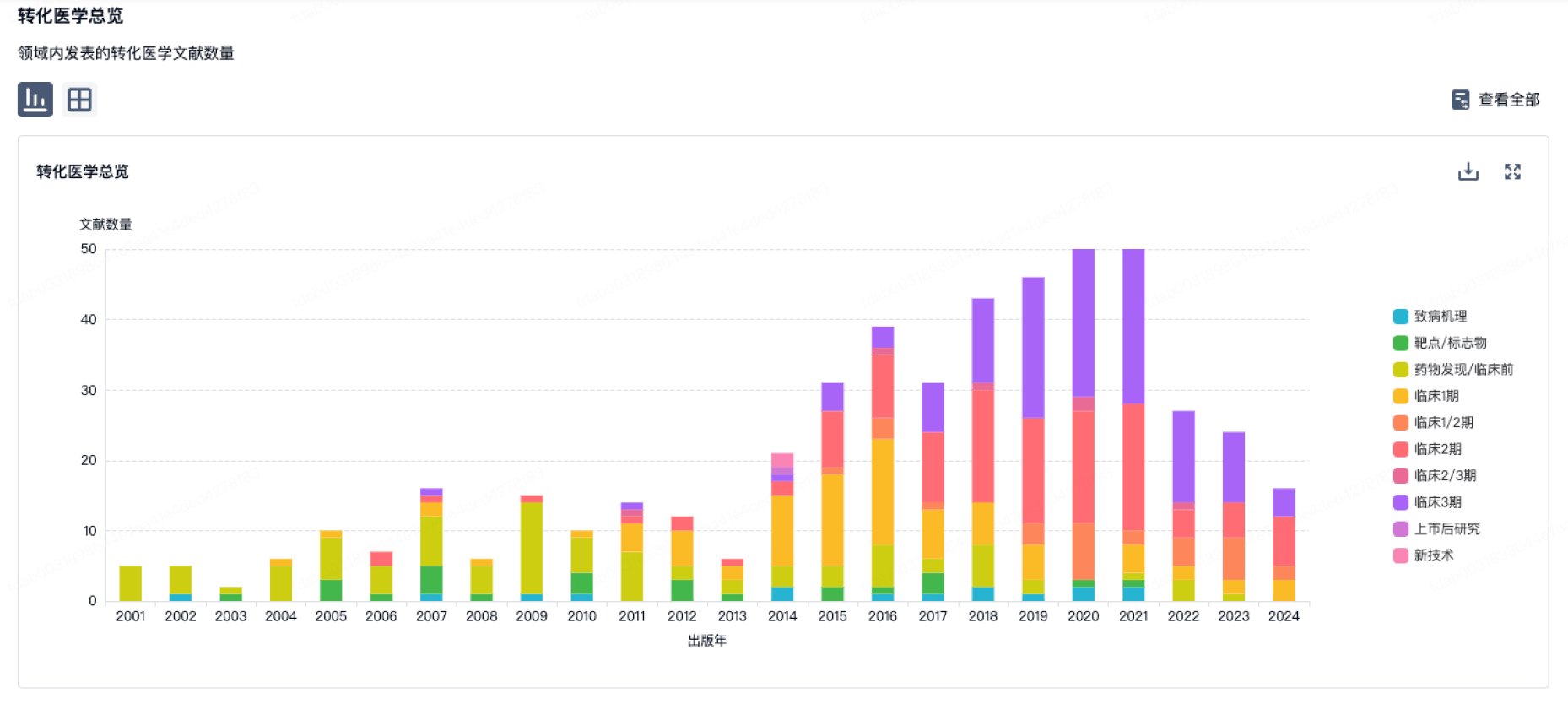
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用