预约演示
更新于:2026-04-12
KYM Biosciences, Inc.
合资公司|2019|United States
合资公司|2019|United States
更新于:2026-04-12
概览
关联
100 项与 KYM Biosciences, Inc. 相关的临床结果
登录后查看更多信息
0 项与 KYM Biosciences, Inc. 相关的专利(医药)
登录后查看更多信息
164
项与 KYM Biosciences, Inc. 相关的新闻(医药)2026-04-02
Positive high-level results from the EMERALD-3 Phase III trial showed AstraZeneca’s Imfinzi (durvalumab) in combination with Imjudo (tremelimumab), lenvatinib and transarterial chemoembolisation (TACE) demonstrated a statistically significant and clinically meaningful improvement in the primary endpoint of progression-free survival (PFS) versus TACE alone for patients with unresectable hepatocellular carcinoma (HCC) eligible for embolisation.
At this interim analysis for overall survival (OS), a key secondary endpoint, this combination also demonstrated a trend toward OS improvement versus TACE alone.
Patients in the investigational arms were treated with the STRIDE regimen (Single Tremelimumab Regular Interval Durvalumab), with or without lenvatinib, before TACE, and then alongside TACE.
Although not formally tested at this time, data for the treatment arm evaluating the STRIDE regimen plus TACE versus TACE alone showed strong trends toward improved PFS and OS. The trial will continue to follow OS and other key secondary endpoints in both investigational arms.
HCC is the most common type of liver cancer.1 In 2026, more than 200,000 patients with HCC will be eligible for embolisation, a standard-of-care procedure that blocks the blood supply to the tumour and can also deliver chemotherapy directly to the liver.2-4 However, most patients who receive embolisation experience disease progression or recurrence within six to ten months.5
Ghassan Abou-Alfa, MD, JD, MBA, PhD(hc), Attending Physician, Professor of Medicine at Memorial Sloan Kettering Cancer Center, and principal investigator in the trial said, “Dual immunotherapy with durvalumab and tremelimumab in the STRIDE regimen represents a meaningful advance for patients with embolisation-eligible liver cancer, who currently lack systemic treatment options to keep their cancer from progressing or recurring, with a trend of improving survival. EMERALD‑3 shows we can now significantly reduce the risk of disease progression with STRIDE as the immunotherapy backbone alongside lenvatinib and TACE.”
Susan Galbraith, Executive Vice President, Oncology Haematology R&D, AstraZeneca, said: “EMERALD‑3 now shows that bringing the dual immunotherapy STRIDE regimen earlier, alongside TACE and lenvatinib, can further improve outcomes in earlier‑stage liver cancer. This builds on the HIMALAYA Phase III trial data in patients with advanced, unresectable disease, where the STRIDE regimen has already demonstrated durable overall survival benefit. We are discussing these positive data with global regulatory authorities while awaiting the final results from the key secondary endpoints.”
The safety profile for each combination was consistent with the known profiles of each medicine, and there were no new safety findings.
These data will be presented at a forthcoming medical meeting and shared with global regulatory authorities.
+++
Notes
Liver cancer Liver cancer, of which HCC is the most common type, is the third-leading cause of cancer death.1,6 In 2026, more than 200,000 patients will be diagnosed with embolisation-eligible HCC.2 Embolisation is a standard-of-care procedure that blocks the blood supply to the tumour and can also deliver chemotherapy directly to the liver.3-4
Immunotherapy is a proven treatment modality in HCC with approved options available for patients in later-line settings.7
EMERALD-3 EMERALD-3 is a randomised, open-label, sponsor-blinded, multicentre, global Phase III trial of a single priming dose of Imjudo 300mg added to Imfinzi 1500mg followed by Imfinzi every four weeks (STRIDE regimen) plus TACE with or without lenvatinib versus TACE alone in a total of 760 patients with unresectable HCC eligible for embolisation.
Participants were randomised in a 1:1:1 ratio to Arm A (TACE, Imfinzi, Imjudo, lenvatinib), Arm B (TACE, Imfinzi, Imjudo) and Arm C (TACE) until each arm reached 175 participants. Randomisation was then continued in a 1:1 ratio to treatment Arms A and C until each reached approximately 275 participants. Patients received Imfinzi with Imjudo, plus TACE as needed, with or without lenvatinib concurrently, followed by Imfinzi with or without lenvatinib until progression.
The trial was conducted in 171 centres across 22 countries, including in North America, Europe, South America and Asia. The primary endpoint is PFS for Imfinzi plus Imjudo, lenvatinib and TACE versus TACE alone. Secondary endpoints include OS for Imfinzi plus Imjudo, lenvatinib and TACE, and PFS and OS for Imfinzi plus Imjudo and TACE versus TACE alone.
Imfinzi Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour's immune-evading tactics and releasing the inhibition of immune responses.
In gastrointestinal (GI) cancer, Imfinzi is approved in combination with chemotherapy in locally advanced or metastatic biliary tract cancer (BTC) and in combination with Imjudo in unresectable HCC. Imfinzi is also approved as a monotherapy in unresectable HCC in Japan and the EU.
In addition to its indications in GI cancers, Imfinzi is the global standard of care based on OS in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiotherapy (CRT). Additionally, Imfinzi is approved as a perioperative treatment in combination with neoadjuvant chemotherapy in resectable NSCLC, and in combination with a short course of Imjudo and chemotherapy for the treatment of metastatic NSCLC. Imfinzi is also approved for limited-stage small cell lung cancer (SCLC) in patients whose disease has not progressed following concurrent platinum-based CRT; and in combination with chemotherapy for the treatment of extensive-stage SCLC.
Perioperative Imfinzi in combination with neoadjuvant chemotherapy is approved in the US, EU, Japan and other countries for patients with muscle-invasive bladder cancer based on results from the NIAGARA Phase III trial. Additionally, in May 2025, Imfinzi added to Bacillus Calmette-Guérin induction and maintenance therapy met the primary endpoint of disease-free survival for patients with high-risk non-muscle-invasive bladder cancer in the POTOMAC Phase III trial.
Imfinzi in combination with chemotherapy followed by Imfinzi monotherapy is approved as a 1st-line treatment for primary advanced or recurrent endometrial cancer (mismatch repair deficient disease only in the US and EU). Imfinzi in combination with chemotherapy followed by Lynparza (olaparib) and Imfinzi is approved for patients with mismatch repair proficient advanced or recurrent endometrial cancer in the EU and Japan.
Since the first approval in May 2017, more than 414,000 patients have been treated with Imfinzi. As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with NSCLC, bladder cancer, breast cancer, ovarian cancer and several GI cancers.
Imjudo Imjudo (tremelimumab) is a human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Imjudo blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death. In addition to its approved indications in liver and lung cancers, Imjudo is being tested in combination with Imfinzi across multiple tumour types including in SCLC (ADRIATIC) and bladder cancer (VOLGA and NILE).
AstraZeneca in GI cancers AstraZeneca has a broad development programme for the treatment of GI cancers across several medicines and a variety of tumour types and stages of disease. In 2022, GI cancers collectively represented approximately 5 million new cancer cases leading to approximately 3.3 million deaths.8
Within this programme, the Company is committed to improving outcomes in gastric, liver, biliary tract, oesophageal, pancreatic and colorectal cancers.
In addition to its indications in BTC and HCC, Imfinzi is being assessed in combinations, including with Imjudo, in oesophageal and gastric cancers in an extensive development programme spanning early to late-stage disease across settings.
Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate (ADC), is approved in the US and several other countries for HER2-positive advanced gastric cancer. Enhertu is jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
Lynparza, a first-in-class PARP inhibitor, is approved in the US and several other countries for the treatment of BRCA-mutated metastatic pancreatic cancer. Lynparza is developed and commercialised in collaboration with MSD (Merck & Co., Inc. inside the US and Canada).
The Company is also assessing rilvegostomig, a PD-1/TIGIT bispecific antibody, in combination with chemotherapy as an adjuvant therapy in BTC, in combination with bevacizumab with or without Imjudo as a 1st-line treatment in patients with advanced HCC, and as a 1st-line treatment in patients with HER2-negative, locally advanced unresectable or metastatic gastric and gastroesophageal junction cancers. Rilvegostomig is also being evaluated in combination with Enhertu in previously untreated, HER2-expressing, locally advanced or metastatic BTC.
AstraZeneca is advancing multiple modalities that provide complementary mechanisms for targeting Claudin 18.2, a promising therapeutic target in gastric cancer. These include sonesitatug vedotin, a potential first-in-class ADC licensed from KYM Biosciences Inc., currently in Phase III development; AZD5863, a novel Claudin 18.2/CD3 T-cell engager bispecific antibody licensed from Harbour Biomed in Phase I development; and AZD4360, an ADC, currently being evaluated in a Phase I/II trial in patients with advanced solid tumours.
In early development, AstraZeneca is developing AZD7003, a Glypican 3 (GPC3) armoured CAR T, in HCC.
AstraZeneca in immuno-oncology (IO) AstraZeneca is a pioneer in introducing the concept of immunotherapy into dedicated clinical areas of high unmet medical need. The Company has a comprehensive and diverse IO portfolio and pipeline anchored in immunotherapies designed to overcome evasion of the anti-tumour immune response and stimulate the body’s immune system to attack tumours.
AstraZeneca strives to redefine cancer care and help transform outcomes for patients with Imfinzi as a monotherapy and in combination with Imjudo as well as other novel immunotherapies and modalities. The Company is also investigating next-generation immunotherapies like bispecific antibodies and therapeutics that harness different aspects of immunity to target cancer, including cell therapy and T-cell engagers.
AstraZeneca is pursuing an innovative clinical strategy to bring IO-based therapies that deliver long-term survival to new settings across a wide range of cancer types. The Company is focused on exploring novel combination approaches to help prevent treatment resistance and drive longer immune responses. With an extensive clinical programme, the Company also champions the use of IO treatment in earlier disease stages, where there is the greatest potential for cure.
AstraZeneca in oncology AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca AstraZeneca (LSE/STO/NYSE: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca’s innovative medicines are sold in more than 125 countries and used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Social Media @AstraZeneca.
Contacts For details on how to contact the Investor Relations Team, please click here. For Media contacts, click here.
References
Dr. Abou-Alfa provides consulting and advisory services to AstraZeneca.
Matthew Bowden Company Secretary AstraZeneca PLC
Oncology Corporate and financial
免疫疗法临床结果临床3期上市批准临床2期
2026-03-26
·证券之星
证券之星消息,近期乐普生物-B(02157.HK)发布2025年年度财务报告,报告中的管理层讨论与分析如下:业务回顾:国内商业化及许可交易 于报告期内,本集团的收入实现显着增加,录得总收入约为人民币934.9百万元,约为2024年收入人民币367.8百万元的2.5倍。 就国内商业化而言,本集团录得销售普佑恒(普特利单抗注射液)及美佑恒(注射用维贝柯妥塔单抗)的收入约人民币501.0百万元,较2024年录得的销售收入显着增加66.8%(2024年:人民币300.3百万元)。同时,于2025年10月获得国家药监局批准的新上市产品美佑恒于报告期内贡献了初步收入,进一步丰富了本集团的商业化产品组合,并为大幅增长奠定坚实基础。 就许可业务而言,本集团已录得收入约人民币424.2百万元(2024年:人民币22.0百万元),主要来自MRG007的对外授权以及TCE资产的对外授权。我们始终致力于推进全球授权战略,并积极开展对外授权合作。于2025年1月,本公司与ArriVent订立独家许可协议,据此,本公司授予ArriVent在大中华区以外地区开发、制造及商业化MRG007的独家权利。根据协议条款,除销售净额的分级特许权使用费外,本公司有资格获得总计最高达12亿美元的首付款、开发、监管及销售里程碑付款。此外,于2025年8月1日,本公司与Excalipoint就本集团自主研发的T细胞衔接器平台TOPAbody开发的两项临床前资产相关的若干知识产权的对外授权及╱或转让订立许可交易。本公司有资格获得合共1,000万美元的现金首付款,外加Excalipoint开曼经扩大已发行股本的10%(将发行予本公司全资附属公司)、总额最高达8.475亿美元的现金开发及商业里程碑付款,以及分级计算的销售特许权使用费。该等交易展现了本公司在全球合作伙伴策略方面日益增长的专业能力,同时在全球寻求战略合作伙伴以在国际市场上推进其管线资产方面持续积累经验。 最值得注意的是,于2025年,我们的全球首创EGFR靶向ADC美佑恒已在中国获得上市批准。同时,我们有更多候选药物进入关键临床阶段,且通过开发及优化联合治疗方案实现了治疗线数前移。于2025年及直至本年度业绩公告日期,本集团候选药物的进展及最新状况描述如下: 美佑恒(注射用维贝柯妥塔单抗) 美佑恒(注射用维贝柯妥塔单抗)是一种由EGFR靶向单抗与强效的微管破坏有效载荷MMAE分子通过vc链接体偶联而成的ADC。其特异性地以高亲和力结合肿瘤细胞表面的人EGFR,在链接体的内化及溶酶体蛋白酶裂解后释放强效的有效载荷,从而导致肿瘤细胞死亡。于2025年10月,美佑恒获得国家药监局批准用于治疗R/MNPC,成为中国首款获批的EGFR靶向ADC。 -NPC:用于治疗R/MNPC的IIb期关键临床研究的良好数据于2025年ASCO大会上作为LBA读出,且以口头汇报形式呈现。截至2024年6月30日,与化疗相比,美佑恒显示出PFS的显着改善,中位PFS分别为5.82个月及2.83个月,疾病进展╱死亡的风险降低37%。此外,美佑恒组的ORR为30.2%,而化疗组为11.5%。截至2024年12月31日,在美佑恒组中已观察到明显的OS改善趋势,两组患者的mOS分别为17.08个月及11.99个月,而mOS尚未成熟。 我们亦正在进行美佑恒(注射用维贝柯妥塔单抗)与普特利单抗联合治疗R/MNPC的III期临床试验。截至2025年4月27日,美佑恒与普特利单抗联合疗法的II期临床试验显示,在先前IO及含铂化疗失败的患者中具有显着且持续的临床获益,确认的客观缓解率(cORR)达到73.3%,确认的疾病控制率(cDCR)达到93.3%,中位无进展生存期(mPFS)为10.9个月;中位总生存期(mOS)尚未达到,12个月及18个月总生存率分别为92.8%及82.5%。该等发现已于2025年ESMO大会上呈现。美佑恒联合普特利单抗于2025年9月获国家药监局CDE授予BTD,用于治疗至少接受过一次含铂化疗及PD/L1抑制剂治疗失败的R/MNPC患者,有望为这一临床需求未得到满足的患者群体提供有效的治疗选择。 -HNSCC:截至2025年12月31日,我们正在进行HNSCC的随机、开放、多中心III期临床研究。 在美佑恒与普特利单抗联合治疗方面,我们目前正在进行HNSCC的II期临床试验(该试验已提线至一线治疗),其II期临床试验的良好数据已于2025年ESMO大会上呈现。截至2025年2月28日,2.0mg/kg剂量组达到4.8%的CR率、47.6%的ORR及95.2%的DCR。对于2.3mg/kg剂量组,ORR及DCR分别为60%及100%。2.0mg/kg剂量组的中位PFS为5.2个月,2.3mg/kg剂量组的中位PFS尚未成熟。我们亦正在欧洲进行针对LA-HNSCC的II期临床试验。此外,我们获得国家药监局CDE关于在中国开展美佑恒与普特利单抗联合疗法的IND批准,旨在评估该方案在手术干预前对术前LA-HNSCC患者的疗效及安全性。 CMG901 CMG901是一种CLDN18.2靶向ADC,由CLDN18.2特异性抗体、可裂解连接子及毒性载荷MMAE组成。其为首个在中国及美国均获得IND批准的CLDN18.2靶向ADC。CLDN18.2于GC、PC及其他实体瘤中呈高度选择性及广泛表达,使其成为治疗开发的理想肿瘤靶点。其由我们与康诺亚透过合营企业KYM共同开发。于2023年2月,AstraZeneca获授予CMG901(AZD0901)的研究、开发、注册、生产及商业化的全球独家许可。截至本公告日期,AstraZeneca已开展多项关于CMG901(AZD0901)治疗晚期实体瘤的临床研究,适应症包括GC、PC及胆道癌。 截至本公告日期,除上述临床试验外,AstraZeneca亦进行了多项sonesitatugvedotin(CMG901/AZD0901)治疗晚期实体瘤的临床研究,针对包括胃癌、胰腺癌及胆道癌的适应症(仅列出处于最高临床阶段的针对相同适应症的试验): (1)一项多中心、开放标签、申办方盲法、随机的III期临床研究,比较AZD0901单药治疗与研究者选择的治疗在伴有CLDN18.2表达的晚期╱转移性胃癌或胃食管连接部腺癌的成人受试者(先前曾接受过二线或以上治疗)(CLARITYGastric01)。 (2)一项Sonesitatugvedotin(AZD0901)联合卡培他滨(伴有或不伴有Rilvegostomig)一线治疗CLDN18.2阳性、HER2阴性、晚期╱转移性胃癌、胃食管连接部癌或食管腺癌的多中心、随机、对照III期临床试验(CLARITY-Gastric02)。2026年2月,该临床试验的首位患者获得给药,并触发了合共45百万美元的里程碑付款。AstraZeneca已作出相应里程碑付款。 (3)一项开放标签、多药物、多中心的II期研究,以评估一种新药或联合疗法作为受试者围手术期治疗局部晚期、可切除胃食管连接部腺癌(GEMINI-PeriOpGC)。 (4)一项II期、开放标签、多中心的临床研究,以评估AZD0901单一疗法及联合抗肿瘤药物治疗CLDN18.2表达晚期实体瘤患者的安全性、耐受性、疗效、药代动力学及免疫原性(包括胃癌╱胃食管连接部腺癌、胰腺癌、胆道癌)(CLARITY-PanTumour01)。 -上市规则第18A.08(3)条规定的警示声明:本公司无法确保本公司将能成功开发及最终成功销售CMG901。股东及我们的潜在投资者在买卖股份时务请审慎行事。 MRG004A MRG004A是一种新型TF靶向位点特异性偶联ADC。我们已在中国完成实体瘤I期临床研究。于2025年8月,我们启动了MRG004A的关键III期临床试验,并于2026年1月完成首例患者入组;同月亦获国家药监局CDE授予BTD认定。PC的Ib期扩展阶段良好数据已于2025年ESMO大会上呈现。截至2025年2月10日,对于先前接受过一线治疗的患者,ORR及DCR分别为40.0%及80.0%,对应的mPFS及mOS分别为5.8个月及13.2个月。对于先前接受过二线或以上治疗的患者,ORR及DCR分别达到18.5%及70.4%,而mPFS及mOS分别为2.7个月及5.8个月。MRG004A有望为胰腺癌患者提供全新的治疗选择。 -上市规则第18A.08(3)条规定的警示声明:本公司无法确保本公司将能成功开发及最终成功销售MRG004A。股东及我们的潜在投资者在买卖股份时务请审慎行事。 CG0070 CG0070是一款用于治疗对BCG无应答膀胱癌患者的溶瘤腺病毒,目前正由我们的美国合作伙伴CGOncology进行一项MRCTIII期临床研究。观察到的最新良好数据已在第26届SUO年会上作为LBA以口头汇报形式呈现。截至2025年9月1日,75.5%的患者在接受CG0070单药治疗后任何时间达到CR。在HRBCG无应答Ta/T1疾病中,CG0070在3个月、6个月及9个月的HG-EFS分别为95.7%、84.6%及80.4%。 我们自CGOncology许可引进CG0070,并获授予在中国内地、香港及澳门开发、制造及商业化CG0070的权利。截至2025年12月31日,我们已在中国完成I期临床试验,并已启动国内关键临床试验的患者入组。对于CG0070与普佑恒(普特利单抗注射液)的联合疗法,我们已获得国家药监局关于其治疗对BCG无应答NMIBC患者的I期试验的IND批准。 此外,CG0070于2025年获国家药监局CDE授予BTD,用于治疗对先前获批疗法复发或难治的BCG无应答膀胱癌患者,该认定体现了CG0070在满足未被满足医疗需求方面的创新性及潜力。 -上市规则第18A.08(3)条规定的警示声明:本公司无法确保本公司将能成功开发及最终成功销售CG0070。股东及我们的潜在投资者在买卖股份时务请审慎行事。 MRG002 MRG002是一种创新性HER2靶向ADC药物,HER2是在许多癌症类型(包括BC、UC及GC/GEJ等)中异常高表达的靶点分子。我们在中国的MRG002临床发展策略旨在实现MRG002对多种常见恶性肿瘤,尤其是BC的二线或更后线全身性治疗的疗效潜力。上述适应症的注册性临床试验正在进行。我们也通过与癌症免疫联合疗法开展临床研究,不断探索MRG002的潜力,旨在面向更多早期患者,并提供更多选择以满足未被满足的医疗需求。 -单一疗法 HER2高表达BC:我们已在中国完成针对伴有肝转移的HER2高表达BC的关键II期临床试验,并观察到良好的数据。截至2025年12月31日,我们正在进行HER2阳性BC的III期临床研究。 -上市规则第18A.08(3)条规定的警示声明:本公司无法确保本公司将能成功开发及最终成功销售MRG002。股东及我们的潜在投资者在买卖股份时务请审慎行事。 MRG001 MRG001是一种临床进度领先的CD20靶向ADC药物,可满足对利妥昔单抗存在原发性耐药或对利妥昔单抗及标准化疗联合治疗存在获得性耐药的B细胞NHL患者的医疗需求。我们已在中国完成MRG001的Ib期剂量扩展研究,并于DLBCL中观察到良好的初步数据。同时,MRG001与BTK抑制剂联合治疗DLBCL患者的II期临床研究正持续进行,其中期数据已于第67届ASH年会上呈现。截至2025年8月31日,在年龄18岁或以上、ECOGPS0-2、经组织学证实为R/RDLBCL且先前至少接受过一线治疗(其中80.8%先前接受过二线或以上全身性治疗,中位数为三线)的可评估患者中,ORR及DCR分别为66.7%及85.7%。该患者群体的中位DoR达到10.2个月,mPFS为13.1个月,且mOS尚未达到。 -上市规则第18A.08(3)条规定的警示声明:本公司无法确保本公司将能成功开发及最终成功销售MRG001。股东及我们的潜在投资者在买卖股份时务请审慎行事。 MRG006A MRG006A是一款基于新型拓扑异构酶I抑制剂且具有全球FIC潜力的GPC-3靶向ADC候选产品,乃基于我们的Hi-TOPiADC平台开发。我们目前正在中国进行HCC的II期临床试验。此外,我们获得FDA的MRG006AIND核准,且该药物获FDA授予FTD及ODD认定。I期临床研究的良好数据已读出,并计划于2026年ASCO大会上呈现。于临床前研究中,MRG006A在多种CDX模型及HCCPDX模型中展现出强大的剂量依赖性的对肿瘤的生长抑制作用。同时,MRG006A亦在探索性毒理学研究中表现出良好的耐受性。 -上市规则第18A.08(3)条规定的警示声明:本公司无法确保本公司将能成功开发及最终成功销售MRG006A。股东及我们的潜在投资者在买卖股份时务请审慎行事。 MRG007 基于临床前及IND支持性研究,MRG007是一款治疗消化道恶性肿瘤的潜在同类最佳ADC。我们目前正在进行一项治疗不可切除的局部晚期或转移性实体瘤的Ia期临床试验。2026年3月,我们的合作伙伴ArriVent已在美国完成了首例患者入组。双方将共同开展全球多中心临床试验(MRCT)。于2025年AACR年会上呈现的MRG007的临床前数据显示出治疗消化道癌的良好临床潜力。 于2025年1月22日,本公司与ArriVent就MRG007的开发及商业化订立独家许可协议。根据协议条款,本公司授予ArriVent在大中华区以外地区开发、制造及商业化MRG007的独家权利。一次性首付款及近期里程碑付款金额为4,700万美元,且本公司有资格获得最高达11.6亿美元的开发、监管及销售里程碑付款,以及在大中华区以外地区销售净额的分级特许权使用费。截至2025年12月31日,我们已收到首付款。 -上市规则第18A.08(3)条规定的警示声明:本公司无法确保本公司将能成功开发及最终成功销售MRG007。股东及我们的潜在投资者在买卖股份时务请审慎行事。 普佑恒(普特利单抗注射液) -普佑恒(普特利单抗注射液)是一种针对人PD-1的人源化IgG4单抗,可拮抗PD-1信号,通过阻断PD-1与其配体PD-L1及PD-L2的结合来恢复免疫细胞杀死癌细胞的能力,并自2022年下半年起已商业化用于治疗MSI-H/dMMR及不可切除或转移性黑色素瘤。于2023年4月,两项适应症获纳入2023年CSCO指南,即普特利单抗作为MSI-H/dMMR结直肠癌及实体瘤的二线或以上治疗,以及普特利单抗作为黑色素瘤的二线治疗。此外,普特利单抗治疗晚期及复发性MSI-H/dMMR妇科癌症获纳入2023年CSGO指南。基于II期研究结果,普特利单抗在MSI-H/dMMR患者中显示出强大的抗肿瘤活性,我们在2025年ASCO年会上呈现了长期生存结果及最新的安全性概况。 oMSI-H/dMMR实体瘤:截至2025年12月31日,我们正在进行一线MSI-H/dMMR转移性结直肠癌的开放标签、多中心及随机III期临床试验,作为附条件上市批准的确证性临床研究。 o黑色素瘤:截至2025年12月31日,我们正在进行针对IV期(M1c)黑色素瘤受试者一线治疗的开放标签、多中心及随机III期临床试验,作为附条件上市批准的确证性临床研究。 创新平台 我们持续致力于建立和开发新型技术平台,将其作为本公司的创新引擎。我们亦已为ADC候选药物开发多个创新的链接体-有效载荷平台,包括Hi-TOPiADC平台及其他早期阶段的平台。于报告期内,我们的创新ADC平台取得了重大进展。依托该等创新平台,我们已开发出两款ADC候选药物,分别为具有全球首创新药潜力的MRG006A及具有全球同类最优潜力的MRG007。两款候选药物均已显示出稳健且可重复的临床前疗效,并具有良好的耐受性安全性概况,且已在中国成功获得IND批准并迅速启动临床。MRG007的临床前数据已于2025年4月在AACR年会上呈现。此外,我们计划于2026年4月的AACR年会上呈现一款双特异性ADC候选药物及一款新型免疫肿瘤融合蛋白候选产品的临床前数据。 -Hi-TOPiADC平台:Hi-TOPiADC平台的特点是:(i)以最佳亲水性设计的链接体,确保强大的可开发性及良好的成药性,其在血液循环中高度稳定并在细胞内高效释放有效载荷;(ii)有效载荷,具有优于竞争对手的良好效力(其并非Pgp的底物,因此具有克服耐药性的巨大潜力);(iii)使用新型链接体-有效载荷的ADC在多种肿瘤类型的PDX中显示出很强的抗肿瘤活性,并显示出良好的安全性概况,且猴子对其表现出良好的耐受性;及(iv)改善治疗窗口。 利用新型链接体-有效载荷平台,我们已开发MRG006A,其为具有全球首创新药潜力的ADC候选药物,目前正在中国进行II期临床试验。 -双特异性ADC:通过利用双特异性ADC技术共同参与靶点A和B,双特异性抗体(BsAb)ADC可以显着扩大包括肺癌、结直肠癌(CRC)及其他癌症在内的关键适应症的治疗范围。 -下一代PD-1:PD-1×细胞因子双特异性抗体旨在克服对现有PD-1疗法的原发性和获得性耐药性。以PD-1+癌症免疫平台为基础,该方法有望显着提高ORR并延长OS。其涵盖广泛的肿瘤类型,且当与ADC结合时或会提供有意义的生存获益,从而转化为对患者有意义的生存获益。 -T细胞衔接器平台:我们的T细胞衔接器平台-TOPAbody-的特点是(i)同时启动TCR信号和共刺激途径,旨在释放T细胞的全部潜能;及(ii)肿瘤微环境中的活性受限。 生产设施 我们在北京的制造厂运营一条2,000L符合GMP标准的生物反应器生产线,其主要支持临床药物供应的生产、提供CDMO生产服务并能够持续优化获批药物的工艺。于报告期内,我们就提供CDMO服务确认收入人民币9.6百万元。 此外,上海生物科技园的生产设施设计总产能为12,000L,且已获得生产单抗及ADC的环境影响评估报告。展望未来,我们将根据ADC候选药物商业化产生的业务需求,继续建设或扩展我们的生产设施。 与乐普医疗的持续关连交易 于2025年11月28日,本公司与乐普医疗就本公司及╱或其附属公司向乐普医疗及╱或其附属公司提供用于开发GLP-1及相关产品的CDMO技术服务订立框架协议。截至2026年12月31日止年度,提供CDMO服务的年度上限为人民币18.2百万元。 于同日,本公司亦与乐普医疗就本集团向乐普医疗及╱或其附属公司及╱或联系人采购临床试验用原辅材料、医药产品、临床试验生物样本检测服务、员工福利产品及其他服务订立另一份框架协议。截至2026年12月31日止年度的年度上限为人民币12.0百万元。 有关上述与乐普医疗持续关连交易的进一步详情,请参阅本公司日期为2025年11月28日的公告。 H股全流通完成 于2025年7月21日,本公司54,268,364股非上市股份转换为本公司H股完成,且该等转换后的H股于2025年7月22日上午九时正开始在联交所上市。详情请参阅本公司日期为2025年7月21日的公告。 采纳限制性股份单位计划 于2025年12月18日,本公司在2025年第二次临时股东大会上获得股东对限制性股份单位计划的批准。限制性股份单位计划旨在通过向本集团合资格雇员及董事(不包括独立非执行董事)授予限制性股份单位来吸引、激励及挽留关键人员,惟须遵守计划规则所载的归属条件、业绩目标及追回机制。股东亦批准了一项上限,限制根据限制性股份单位计划及任何其他股份激励计划可能发行的新股份总数不得超过本公司股份总数(不包括库存股份,如有)的5%。业务展望:本公司是一家聚焦于肿瘤治疗领域的创新型生物制药企业,致力于推动中国创新型ADC的技术进步,以更好地填补癌症患者的临床需求缺口。展望2026年,我们计划通过以下发展策略充分发挥我们的竞争优势: 在药物研发方面,我们致力于将自主研发能力与战略合作相结合,聚焦临床需求显着的适应症领域,不断丰富差异化的商业化产品组合。我们将进一步聚焦于推动下一代ADC药物及免疫肿瘤双╱三特异性抗体的战略研发,同时加快后期产品的商业化。此外,我们的核心候选药物正进入关键临床阶段。MRG004A目前正在进行III期临床试验的患者入组。此外,我们已启动CG0070国内关键临床试验的患者入组。我们亦将加快推进包括MRG006A及MRG007在内的其他创新候选药物进入关键临床阶段。同时,我们正在不断探索管线内联合疗法的潜在疗效,努力为更广泛的患者群体带来更大的临床效益。 在国内商业化方面,我们已通过自有销售渠道成功商业化普佑恒(普特利单抗注射液),充分验证我们的销售策略和商业模式。此外,对于美佑恒(注射用维贝柯妥塔单抗),国家药监局已在中国授予上市批准。我们将继续集中资源及努力推动商业化进程,专注于提高我们获批产品的市场占有率及销售表现。我们将采取进一步措施提高这两款产品的市场可及性,加快各级市场渗透,进一步扩大市场份额。凭藉我们商业化团队的专业知识及行业资源,我们将寻求通过市场营销、学术推广等各种方式提升我们的品牌形象及市场认知度。同时,我们将根据美佑恒(注射用维贝柯妥塔单抗)的实际销售表现优化商业化策略,并利用我们已全面扩大的营销及商业化团队,专注于推动总收入增长。我们相信加强市场拓展力度能够有效提升市场准入能力、扩大市场份额,并增加我们已商业化产品的销售额和品牌影响力,从而为我们候选药物的未来商业化奠定坚实的市场和渠道基础。 在国际方面,我们将加大在全球市场的拓展力度。我们的ADC平台已获跨国公司认可-CMG901的全球权利已成功对外授权予AstraZeneca,MRG007的大中华区以外地区权利已对外授权予ArriVent。我们预期候选药物将迎来更广阔的业务开发机遇。展望未来,我们将坚持不懈地扩大我们的国际网络,并探索新的业务开发合作机会。我们将继续致力于在全球范围内寻找更多战略合作伙伴,通过合作、授权协议或合营企业等方式开发我们的ADC产品和其他创新候选药物。以上内容为证券之星据公开信息整理,由AI算法生成(网信算备310104345710301240019号),不构成投资建议。
引进/卖出抗体药物偶联物财报
2026-03-24
·药事纵横
3月初,康诺亚宣布核心产品CMG901(Claudin18.2 ADC)在全球授权合作伙伴AstraZeneca 的推动下,已启动一项联合卡培他滨联合或不联合Rilvegostomig一线治疗Claudin18.2阳性、HER2阴性的晚期/转移性胃癌、胃食管结合部癌或食管腺癌的多中心、随机对照、III期临床研究(CLARITY-Gastric 02),并完成了首例受试者给药。根据许可协议条款及条件,该研究进展触发总金额4500万美元里程碑付款,目前,AstraZeneca支付的该笔里程碑付款已到账。
随着CMG901触发里程碑付款,司普奇拜单抗于2025年底成功纳入国家药品医保目录,康诺亚的商业化和出海路径均已成功打通。随着CM336、CM518D1、CM512等产品数据的逐步成熟,康诺亚有望进一步链接研发和商业化,由Biotech向BioPharma华丽转型。
图:康诺亚产品管线
数据来源:康诺亚宣传资料
(一)司普奇拜单抗:成功纳入国家医保目录,商业化放量在即
司普奇拜单抗是首款上市的国产IL-4抑制剂,目前已获批三项适应症,分别是特应性皮炎、慢性鼻窦炎伴鼻息肉和过敏性鼻炎。2025年12月,司普奇拜单抗三项适应症均成功纳入2025年国家基本医疗保险药品目录,有望进一步提升司普奇拜单抗的可及性和可负担性,开启司普奇拜单抗商业化腾飞的元年。
司普奇拜单抗在中重度特应性皮炎治疗中展现出快速、强效、持久清除皮损、头颈部应答更快更强的治疗优势。单药治疗52周的EASI-75(湿疹面积及严重程度指数评分较基线降低≥75%)和EASI-90(湿疹面积及严重程度指数评分较基线降低≥90%)应答率分别高达92.5%和77.1%,对未接受过其他系统治疗的患者疗效更优,EASI-75及EASI-90应答率可达95.4%和79.2%。与全球首个IL-4单抗度普利尤单抗相比,度普利尤单抗治疗特应性皮炎52周的EASI-75仅65%,司普奇拜单抗优势十分明显。
2025年上半年,司普奇拜单抗销售额为1.7亿元,根据国海证券研究报告,随着司普奇拜单抗正式纳入医保报销范围,预计2026年公司收入达7.4亿元。基于司普奇拜单抗填补多项适应症的治疗空白,包括慢性鼻窦炎伴鼻息肉和季节性过敏性鼻炎等适应症,司普奇拜单抗未来峰值销售额有望达50亿元。
图:司普奇拜单抗峰值有望达50亿元
数据来源:康诺亚宣传资料
(二)CMG901:4500万美元里程碑付款夯实信心,收获期将至
CMG901是由康诺亚和上海美雅珂共同开发的新型靶向Claudin18.2的全新重组人源化单克隆抗体偶联药物,目前处于临床Ⅲ期。CMG901可通过多种机理杀伤肿瘤细胞,一方面,CMG901可通过Claudin18.2特异结合Claudin18.2阳性细胞,继而被内吞进入细胞溶酶体。溶酶体中的蛋白酶将链接体降解后会释放微管抑制剂MMAE,导致肿瘤细胞的细胞周期停滞并诱发细胞凋亡。另一方面,CMG901的抗体部分为人IgG1亚型,还可通过ADCC和CDC效应杀伤Claudin18.2阳性细胞。
CMG901兼具疗效和安全性,针对末线胃癌mOS达11.8m。在2024年ASCO上,康诺亚披露了CMG901的Ⅰ期临床数据。三个剂量组2.2mg/kg、2.6mg/kg、3.0mg/kg组共纳入113例胃癌/胃食管结合部腺癌患者(分别为44、50、19例)。受试者既往中位治疗线数为2线,74%的受试者既往接受过抗PD-1/PD-L1治疗。CMG901在三个剂量组的确认的客观缓解率(ORR)为35%,确认的疾病控制率(DCR)为70%。在2.2mg/kg剂量组中观察到的确认的ORR为48%。所有93例Claudin 18.2高表达胃癌/胃食管结合部腺癌受试者的中位无进展期(mPFS)为4.8个月,中位总生存期(mOS)为11.8个月。
图:CMG901治疗2L及以上胃癌数据优异
数据来源:康诺亚宣传资料
2023年2月,康诺亚和乐普生物共同宣布与阿斯利康就 CMG901 达成全球独家授权协议,基于合作条款,由康诺亚和乐普生物合资设立的KYM Biosciences Inc. 获得6300万美元的预付款和超过11亿美元的潜在额外研发和销售相关的里程碑付款,以及高达低双位数的分层特许权使用费。
由于Claudin18.2 ADC的研发候选药物较多,因此市场一度不看好这笔合作授权,甚至担心退货的风险。此次康诺亚/乐普生物收到阿斯利康4500万美元的里程碑付款,无疑是一针强心剂,不但印证了阿斯利康将不遗余力推进CMG901的临床研究,更将向1L胃癌的适应症发起冲锋,彻底打消了市场的担忧。鉴于CMG901的末线胃癌适应症有望于2026年申报上市,CMG901收获期将至。(三)潜力管线:纷至沓来,深挖BIC管线价值
放眼康诺亚产品管线,CM336、CM518D1、CM512等管线均有望掀起新一轮研发浪潮,兼具FIC和BIC的潜力。
(1)CM336是一种BCMAxCD3双特异性抗体,可同时靶向识别并特异性结合靶细胞表面的BCMA和T细胞表面CD3受体,将免疫T细胞招募至靶细胞周围,诱导T细胞介导的细胞杀伤(TDCC)作用杀伤靶细胞。2024年11月,康诺亚与Ouro Medicines Ltd 订立独家许可协议,授予Ouro Medicines在全球研究、开发、生产、注册及商业化CM336的独家权利。
CM336临床数据优异,潜在同类最佳。在II期剂量扩展阶段,仅4.7%受试者发生2级CRS事件,未发生ICANS事件,目标剂量组ORR为95.2%,≥CR率为76.2%,MRD阴性率为100%,12个月PFS无事件率为95.2%。
图:CM336潜在同类最佳,目标剂量组ORR为95.2%
数据来源:康诺亚宣传资料
(2)CM518D1是由康诺亚自主研发的CDH17抗体偶联药物,目前正在中国开展针对实体瘤的I/II期临床研究。临床前研究表明,CM518D1具有较强的直接细胞毒杀伤作用和“旁观者效应”,具备良好的血浆稳定性;CM518D1在多种实体瘤动物模型中展现出优异的抗肿瘤活性;在毒理评价中,CM518D1也展现了优异的安全性和治疗窗口。
CDH17在结直肠癌、胃癌、食管癌、胰腺癌等多种消化道肿瘤高表达,正常组织上CDH17仅表达于肠上皮细胞之间紧密连接的侧膜,因此脱靶毒性低,是肿瘤治疗中十分有潜力的靶点。基于此,多家企业的CDH17 ADC已经成功授权MNC,如翰森制药的HS-20110授权罗氏,映恩生物的DB-1324授权GSK。放眼未来,CM518D1有望凭借优异的临床数据实现对外授权。
图:CM518D1作用机制
数据来源:康诺亚宣传资料
(3)CM512是全球首款长效(TSLP x IL-13)双阻断剂,目前处于临床Ⅱ期,正在开发的适应症包括中重度特应性皮炎、CRSwNP、哮喘、COPD、慢性自发性荨麻疹等多项自身免疫病。
2024年7月,康诺亚宣布将2款双抗新药CM512、CM536的大中华区外全球权益授权给Belenos Biosciences。根据协议,康诺亚将收到1500万美元预付款、1.7亿美元里程碑金额,以及特定比例的销售分成。此外,康诺亚全资附属公司一桥香港将获得Belenos约30.01%的股权。
目前,全球仅三款TSLP x IL-13抑制剂处于临床阶段,分别是赛诺菲的Lunsekimig、康诺亚的CM512和荃信生物的QX027N。与Lunsekimig相比,CM512药物半衰期约70天,优于赛诺菲的Lunsekimig(约10天),免疫原性更低,治疗窗口更大。
图:TSLP/IL-13双抗竞争格局
数据来源:公开数据整理
(四)小结
康诺亚正站在研发和商业化的关键节点,整装待发。从商业化的角度,2026年是司普奇拜单抗纳入国家医保目录的首年,放量在即,剑指50亿元峰值销售额;从研发的角度,CMG901收到阿斯利康4500万美元的里程碑付款,标志着阿斯利康开发的决心;CM336、CM518D1、CM512等潜力管线纷至沓来,接棒下一轮研发浪潮。期待康诺亚在新的一年,以崭新的姿态平衡研发和商业化,从Biotech向Biopharma转型。
数据来源:康诺亚宣传资料
国海证券,《康悦达开出首批医保处方,看好后续医保放量》,20
立即扫码加入药事纵横交流群
100 项与 KYM Biosciences, Inc. 相关的药物交易
登录后查看更多信息
100 项与 KYM Biosciences, Inc. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月04日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
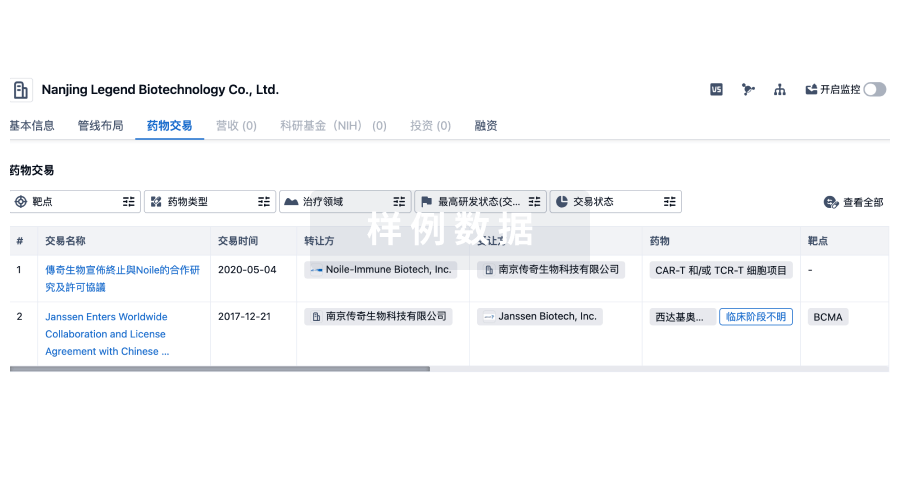
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
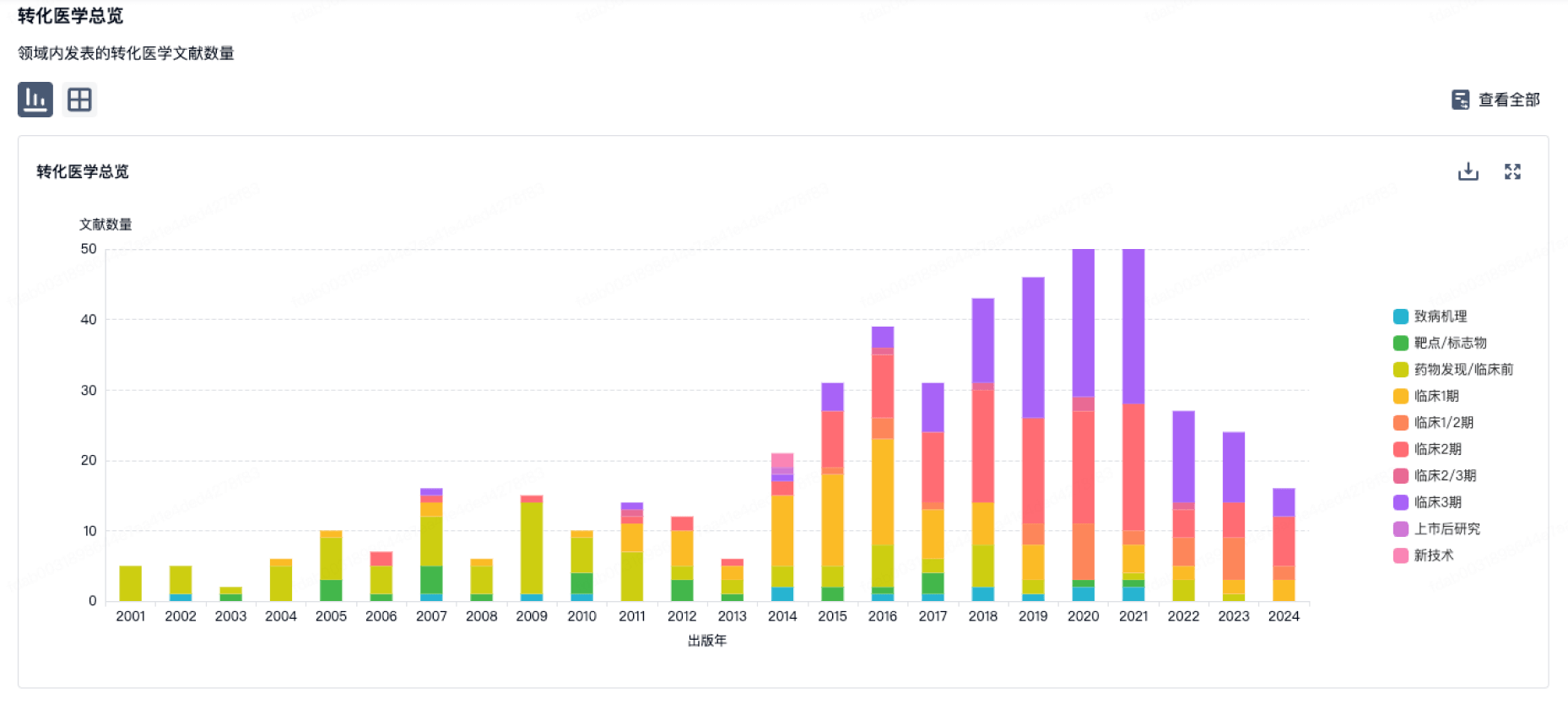
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
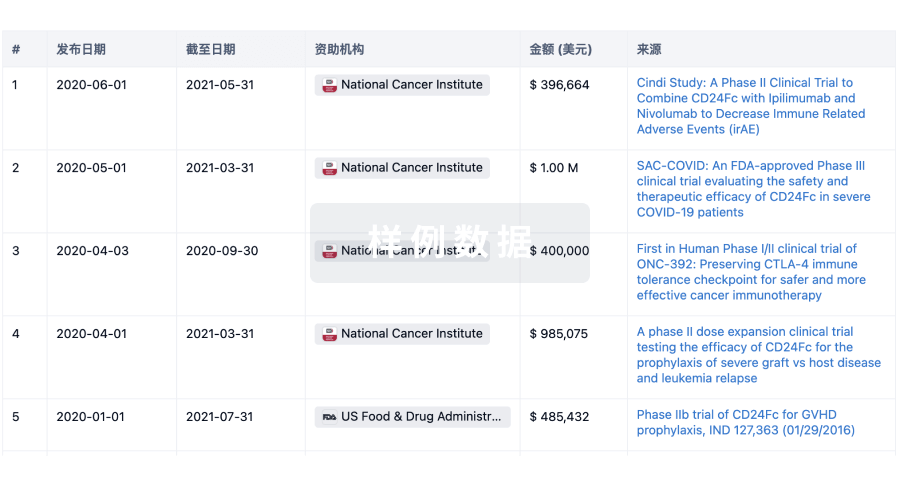
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用