预约演示
更新于:2025-05-07

MidAmerica Health, Inc.
更新于:2025-05-07
概览
关联
100 项与 MidAmerica Health, Inc. 相关的临床结果
登录后查看更多信息
0 项与 MidAmerica Health, Inc. 相关的专利(医药)
登录后查看更多信息
1
项与 MidAmerica Health, Inc. 相关的文献(医药)Journal of Family Psychology3区 · 心理学
Collateral benefits of the family check-up in early childhood: Primary caregivers' social support and relationship satisfaction.
3区 · 心理学
Article
作者: Shaw, Daniel S ; Dishion, Thomas J ; Fosco, Gregory M ; McEachern, Amber D ; Gardner, Frances ; Wilson, Melvin N
80
项与 MidAmerica Health, Inc. 相关的新闻(医药)2025-03-26
关注并星标CPHI制药在线
2025年3月25日,国家药监局发布了2份公告,分别是《国家药监局国家卫生健康委关于颁布2025年版<中华人民共和国药典>的公告》(2025年第29号)和《国家药监局关于实施2025年版<中华人民共和国药典>有关事宜的公告》(2025年第32号)。虽然从文本看,这2份公告和过去历次实施公告保持连贯性,并和现行法规保持一致;但是如果仔细阅读每一条内容,还是可以看出其中某些条款规定是不合理的,甚至是很难实施的。
为了行文顺畅,并便于读者清晰阅读,本文内容展开次序采用和32号公告的内容一致的次序来展开。
公告原文:2025年版《中华人民共和国药典》(以下简称《中国药典》)已由国家药监局、国家卫生健康委2025年第29号公告颁布,自2025年10月1日起实施。现就实施本版《中国药典》有关事宜公告如下:
分析:
——药典开始日期是2025年10月1日,这是毫无疑问的。也就是说,从法律意义上讲,在2025年10月1日开始,MAH签字放行的药品(含原料药),需要符合2025版《中国药典》;为了实现这个目标,MAH需要提前和供应商沟通,在这个日期之前,采购符合2025版《中国药典》的原辅包。
——对于MAH在库未使用完毕的物料,应该采用技术评估手段,确保从2025年10月1日开始,投入生产的各类物料符合2025版《中国药典》。
公告原文:一、根据《药品管理法》规定,药品应当符合国家药品标准。《中国药典》是国家药品标准的重要组成部分,是药品研制、生产(进口)、经营、使用和监督管理等相关单位均应当遵循的法定技术标准。
分析:
——根据上面这段描述,如果生产环节也遵守2025版《中国药典》,是否生产、工程、物料储存环节产生的数据修约工作也需要符合“四舍六入五成双”的原则?
——根据上面这段描述,如果进口药品也遵守2025版《中国药典》,是否国外药品生产、检验产生的数据修约也需要符合中国药典凡例提到的“四舍六入五成双”的原则?那些计算机化系统内置函数是否也必须符合这个修约规则?估计国家药监局和药典委没有考虑这些细节。
公告原文:二、《中国药典》主要包括凡例、品种正文、通用技术要求和指导原则。自实施之日起,所有药品上市许可持有人及生产上市的药品应当执行本公告和本版《中国药典》相关要求。其中,指导原则相关要求为推荐技术要求。
分析:这条内容很好,在描述药典组成部分的同时,还提醒所有药典用户,中国药典部分内容不是强制的,是推荐性技术要求。
公告原文:三、自实施之日起,凡原收载于历版药典、局(部)颁标准的品种,本版《中国药典》收载的,相应历版药典、局(部)颁标准同时废止;本版《中国药典》未收载的,仍执行相应历版药典、局(部)颁标准,但应当符合本版《中国药典》的相关通用技术要求。经上市后评价撤销或者注销的品种,相应历版药典、局(部)颁标准废止。
本版《中国药典》品种正文未收载的制剂规格、中药的制法,其质量标准按本版《中国药典》同品种相关要求执行,规格项、制法项分别按原批准证明文件执行。
分析:
——这条内容很多,争论也不少。笔者坚持认为:这段第一句部分内容是错误的,因为2025版《中国药典》只能取代2020版中国药典+2022年第一增补本;而不能简单地认为中国药典生效后,收载相同品种的局(部)颁标准也会作废。因为这些局(部)颁标准所收载品种可能和药典收载品种采用了不同的生产工艺或者不同的处方,因而,2025版《中国药典》可能不适用于这些品种。
——这段内容的第二句,也存在逻辑问题。试问,如果《中国药典》品种正文收载了某些中药制剂的制法,难道生产这些品种的MAH就必须放弃自己工艺,采用药典上面的制法吗?2025年1月1日生效的《中药标准管理专门规定》第四十条已经提到,中药标准中的制法是概括性表述,不是生产工艺。国家局实施公告,应该对这个问题补充说明,避免歧义。
公告原文:四、本版《中国药典》颁布后,执行药品注册标准的,药品上市许可持有人应当及时开展相关对比研究工作,评估药品注册标准是否符合新颁布的药典标准有关要求。
对于需要变更药品注册标准的,药品上市许可持有人应当在本版《中国药典》实施之日前,按照药品上市后变更管理相关规定提出补充申请、备案或者报告,并按要求执行。
药品注册标准中收载检验项目多于或者异于药典规定的,或者质量指标严于药典要求的,应当在执行药典要求的基础上,同时执行注册标准的相应项目和指标。药品注册标准收载检验项目少于药典规定或者质量指标低于药典要求的,应当执行药典规定。
分析:
——这段第一句,内容很好,MAH要评估药典标准的适用性。这个要求和2024年1月1日生效的《药品标准管理办法》的原则是一致的。
——这段第二句,内容虽然和2024年1月1日生效的《药品标准管理办法》的原则是一致的,但是写得不好。简单说,在现实中,很难执行。如果读者稍微认真读过2021年发布的已上市中药、化药、生物制品药学变更指导原则,都应该知道,在目前变更管控体系下,涉及质量标准的变更没有微小变更这个等级;因此质量标准变更采取的途径或者是补充申请,或者是备案。
那么问题就来了,大部分省局估计没有能力和担当来对企业提出涉及质量标准备案的申请进行处理。这样一来,大部分企业只能采用补充申请方式,试问,国家局审评中心做好这样的工作准备了吗?
——这段第二句的不合理之处还在于,如果辅料和包材企业要变更自己的产品质量标准,因为辅料和包材在目前中国法规体系下是非药品身份,没有合适提交途径。
——针对这段第三句,猛地一看很合理,仔细思考还是存在漏洞和歧义。参见下面表格:
按照这段第三句的表面要求,企业在2025年10月1日执行的质量标准,就应该是12项检测内容。这样要求合理科学吗?
正确理解应该是这样的:MAH必须充分、全面、科学评估药典内容对自己标准是否适用(这个单词非常重要);如果适用,MAH自然要采用药典内容;如果评估结果是药典某些项目不适用于企业产品(由于不同的生产工艺/不同的处方等原因),企业不应该执行这些不适用的项目。
不能逼着企业做简单数学加法,这是错误逻辑!
MAH必须规范管理这些变更评估报告,在自己体系内供官方随时检查。
公告原文:五、为符合本版《中国药典》要求,如涉及药品处方、生产工艺和原料、辅料、直接接触药品的包装材料和容器等变更的,药品上市许可持有人、生产企业应当按照《药品注册管理办法》《药品上市后变更管理办法(试行)》以及有关变更研究技术指导原则和药品生产质量管理规范等要求进行充分研究和验证,按相应变更类别批准、备案后实施或者报告。
分析:这条内容逻辑上、法理上没有问题,但是现实执行上存在很大难度。2025版《中国药典》实施会影响很多企业和很多品种,在短短六个月内完成这些变更,估计药监系统会承受巨大压力,真的可以按照这个公告要求规范执行吗?
另外,如果辅料企业为了符合药典四部最新标准,变更工艺如何办?
公告原文:六、由于溶出度、释放度等项目在质量控制中的特殊性,按照仿制药质量和疗效一致性评价要求核准的仿制药注册标准中有别于《中国药典》的,按经核准的药品注册标准执行。
分析:这条内容很好,和2020版药典实施公告基本保持一致,也和基本事实保持一致。这条也提醒所有企业和药监局:即使都生产同一品种,由于处方和工艺的差异,不同企业的产品会执行不同的质量标准。
公告原文:七、本版《中国药典》已进行通用名称修订的药品,应当使用本版《中国药典》中载明的名称,其原名称可作为曾用名过渡使用。在下一版药典实施之日前,曾用名可与本版《中国药典》中载明的名称同时使用。
分析:这条内容清晰,无歧义,不需要解释。
公告原文:八、自本版《中国药典》实施之日起,提出的药品注册申请,相应申报资料应当符合本版《中国药典》相关要求。
在本版《中国药典》实施之日前已受理,并且尚未完成技术审评的注册申请,自本版《中国药典》实施之日起药品监督管理部门应当按照本版《中国药典》相关要求开展相应审评审批,申请人需要补充技术资料的,应当一次性完成提交。
在本版《中国药典》颁布之日后、实施之日前按原药典标准相关要求批准上市的药品,批准后6个月内应当符合本版《中国药典》相关要求。
分析:这条内容很好,逻辑清晰,滴水不漏。然而现实中执行情况,就看工作人员理解能力和企业配合态度了。
公告原文:九、药品上市许可持有人、生产企业和药品注册申请人应当积极做好执行本版《中国药典》的准备工作,对在《中国药典》执行过程中发现的问题及时向国家药典委员会报告,同时应当持续研究完善药品质量标准,不断提升药品质量控制水平。
分析:药典委需要尽快公布电话和邮箱,不然MAH如何反馈自己发现的问题?
公告原文:十、各省级药品监督管理部门应当配合做好本版《中国药典》的宣传贯彻,加强本版药典执行中的监督与指导,及时收集和反馈相关问题和意见。
分析:希望各省局和各市局、区局积极收集问题,及时反馈。
公告原文:十一、国家药典委员会负责组织和协调本版《中国药典》的宣贯培训和技术指导工作,在官方网站开设“《中国药典》执行专栏”,及时答复执行中反映的问题。
分析:这条内容很好。希望药典委在官网专栏信息更新速度更快,内容更全面。
作者简介
zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。
END
2025金笔奖征文活动开启,来投稿吧!
领取CPHI & PMEC China 2025展会门票
智药研习社直播预告
来源:CPHI制药在线
声明:本文仅代表作者观点,并不代表制药在线立场。本网站内容仅出于传递更多信息之目的。如需转载,请务必注明文章来源和作者。
投稿邮箱:Kelly.Xiao@imsinoexpo.com
▼更多制药资讯,请关注CPHI制药在线▼
点击阅读原文,进入智药研习社~
带量采购核酸药物
2025-01-31
·汇聚南药
转自:企业公告 整理:清风
2025年01月24日,振东制药发布公告,根据裁决结果山西振东制药股份有限公司向北京朗迪制药有限公司支付人民币5亿元,至此国内MAH制度实施以来最大的B证与C证的官司正式落地。
本次仲裁情况:
2024年12月31日,本案在北京进行 了开庭审理。朗迪公司、振东制药均委派了仲裁代理人参加了庭审,该仲裁案件源于2021年朗迪制药与振东制药签订的十年《药品委托生产合同》。
2025年1月24日,公司收到仲裁委员会仲裁庭送达的裁决书,经仲裁委员会调解,仲裁双方当事人达成和解,案件所处的仲裁阶段终局裁决。
根据《仲裁法》等相关规定,仲裁庭经合议作出裁决,裁决摘要如下:
本公司在2025年1月20日后15个工作日内向朗迪公 司支付人民币1亿元。
本公司在2025年1月20日后30个工作日内向朗迪公 司支付人民币2亿元。
本公司在2025年1月20日后60个工作日内向朗迪公 司支付人民币1亿元。
本公司在2025年1月20日后6个月内,且在满足相关条件的情况下向朗迪公司全资子公司支付人民币1亿元。
本公司支付完毕全部款项后,双方之间与《药品委托生 产合同》及其补充合同相关的所有争议全部解决,《股权出售协议》相关所有争议亦全部解决,任何一方不得再以任何理由就前述争议向对方提起诉讼/仲裁或其他权利主张。
双方在本案中支出的、与本案相关的全部费用,包括但不限于律师费、仲裁费、财产保全费、担保费等费用均各自承担。该裁决为终局裁决,自作出之日起生效。
MAH曾申请索赔近15亿
2024年09月09日,振东制药发布公告称,北京朗迪制药有限公司向中国国际经济贸易仲裁委员会申请仲裁振东制药,索赔金额高达14.67亿人民币。其仲裁请求还包括申请仲裁振东制药支付其为实现本案债权所支出的律师费、财产保全费、财产保全担保费,并申请仲裁本案仲裁费由振东制药承担。
该仲裁案件源于2021年朗迪制药与振东制药签订的《药品委托生产合同》。根据双方签订的《药品委托生产合同》,朗迪制药委托振东制药生产碳酸钙D3片(II)、碳酸钙D3颗粒产品,有效期为十年。
朗迪制药认为振东制药在委托生产过程中可能存在违约行为,从而引发了此次合同纠纷。
具体信息详见:14.67亿!B证向C证索赔
MAH生产劣药被罚1.4亿
2023年朗迪制药因生产销售不合格药品收到1.4亿元顶格罚单,这些不合格药品是2021年2月3日—2022年11月29日期间自行生产及委托振东制药生产的的32批次碳酸钙D3颗粒、碳酸钙D3颗粒(Ⅱ)、碳酸钙D3片(Ⅱ)含量测定项下维生素D3不符合规定。
标示为北京振东朗迪制药有限公司、北京振东朗迪制药有限公司委托山西振东制药股份有限公司、北京振东康远制药有限公司委托山西振东制药股份有限公司生产的24批次碳酸钙D3颗粒不符合规定,不符合规定项目均为含量测定。
标示为北京朗迪制药有限公司委托山西振东制药股份有限公司生产的7批次碳酸钙D3颗粒(Ⅱ)不符合规定,不符合规定项目均为含量测定。
标示为北京朗迪制药有限公司委托山西振东制药股份有限公司生产的1批次碳酸钙D3片(Ⅱ)不符合规定,不符合规定项目为含量测定。
北京市市场监督管理局指出,上述32批次不合格药品共计生产931669盒,其中成品留样242盒,成品入库931427盒。至抽检时入库成品已全部售出。截至2023年7月20日,当事人累计召回不合格药品54249盒。
朗迪制药被北京市市场监督管理局责令停产停业整顿30天,并罚款1.34亿余元、没收违法所得618万余元及碳酸钙5.4万余盒。
具体信息详见:朗迪制药被罚1.4亿!32批次含量不合格,原因是→
两家的聚散离合
2016年5月,振东制药作价26.46亿元,溢价11倍收购了朗迪制药100%股权,该公司后更名为朗迪制药。
2016年和2017年,振东制药分别实现营业收入32.83亿元、37.32亿元,净利润2.03亿元、3.02亿元。
尽管2018年受商誉减值因素拖累亏损1.47亿元,但2019年和2020年,振东制药的营业收入继续增长至43.99元、48.48亿元,净利润分别为1.43亿元、2.62亿元。
2021年8月,振东制药以58亿元的价格出售全资子公司朗迪制药100%股权给上海方朗。
短短5年时间,振东制药将朗迪制药买入又卖出,净赚22亿的差价,同时持有期内朗迪制药还贡献了10亿左右的净利润。
虽然100%地出售了朗迪制药的股权,但是振东制药并未完全和朗迪制药脱离联系,二者仍然有密切的业务合作。
2021年8月17日,朗迪制药与振东制药签署《药品委托生产合同》,约定朗迪制药委托振东制药生产碳酸钙D3片(Ⅱ)、碳酸钙D3颗粒产品,委托期限自《股权出售协议》所述交割日起生效,有效期为十年。
值得注意的是,朗迪制药官方发布的召回产品批次显示,最早的生产日期可追溯到2021年2月。这也意味着当时振东制药与朗迪制药仍为母子公司关系。
喜欢我们文章的朋友点个“在看”和“赞”吧,不然微信推送规则改变,有可能每天都会错过我们哦~
免责声明
“汇聚南药”公众号所转载文章来源于其他公众号平台,主要目的在于分享行业相关知识,传递当前最新资讯。图片、文章版权均属于原作者所有,如有侵权,请在留言栏及时告知,我们会在24小时内删除相关信息。
信息来源:蒲公英Ouryao
往期推荐
本平台不对转载文章的观点负责,文章所包含内容的准确性、可靠性或完整性提供任何明示暗示的保证。
财报高管变更并购
2025-01-20
关注并星标CPHI制药在线
FDA作为全球领先的药品审评和监管机构,其工作计划对于医药行业从业者会产生明显影响,因此也为行业所关注。每年,FDA会发布新一年的技术指南修订和制订计划,来说明自己的工作重点。本文将结合2025年FDA CBER指南工作计划和CDER指南工作计划来介绍,希望可以为中国制药行业提供参考。
第一部分:CBER 2025年指南工作计划整体情况
近期,FDA官网发布《Guidance Agenda: Guidance Documents CBER is Planning to Publish During Calendar Year 2025》,对FDA下属CDER将在2025年发布的指南,进行了介绍。详情参见下表。
第二部分:CBER 2025年指南工作计划重点介绍
下面对这份2025年技术指南工作计划,选择重点内容进行介绍。
◆Blood and Blood Components
※Recommendations for the Evaluation of Blood Collection, Processing, and Storage Devices Using Non-Di(2-ethylhexyl) Phthalate (non-DEHP) Materials
翻译:使用非邻苯二甲酸二(2-乙基己基)酯(非DEHP)材料的血液采集、处理和储存器械评估建议。
血液制品是高风险产品,其所使用的包材或者容器材质对于质量影响很关键。因此,对于企业采用新材质,都需要进行充分评估。这份指南将提供评估思路。
◆Therapeutic Products
※Frequently Asked Questions — Cell and Gene Therapy Products
翻译:细胞和基因治疗产品常见问题
细胞和基因治疗产品是这几年国际和美国国内研发热点,有很多问题需要厘清。FDA发布这份指南,将促进相关问题的认知深化。这份指南编制工作在2024年已经被列为工作计划之一,没有完成,这次仍然被列为2025年工作计划之中。
※Post Approval Methods to Capture Safety and Efficacy Data for Cell and Gene Therapy Products
翻译:用于获取细胞和基因治疗产品的安全性和有效性数据的批准后方法
细胞和基因治疗产品属于新型药品,临床使用情况比较特殊。在这类药品获得批准上市后,MAH如何规范收集安全和有效数据,需要官方更明确要求。这份指南将解决这类问题。
第三部分:CDER2025年指南工作计划整体情况
近期,FDA官网发布《CDER Guidance Agenda
New and Revised Draft Guidances Planned for Publication in Calendar Year 2025》,对FDA下属CDER将在2025年发布的指南,进行了介绍。详情参见下表。
分析:和过去历年CDER工作计划安排类似,FDA CDER在2025年技术指南修订和制订工作计划中,重点工作领域还是临床医学、仿制药、ICH专题和CMC专题等。
第四部分:CDER2025年指南工作计划重点介绍
下面对这份2025年技术指南工作计划,选择重点内容进行介绍。
◆Administrative/Procedural专题
※Civil Monetary Penalties for Failure to Meet Accelerated Post Marketing Requirements
翻译:未能满足加速上市后要求的民事罚款
FDA为了促进创新药上市,设置了优先审评程序。但是这些通过优先审评程序上市的药品,需要按照官方要求完成上市后部分补充研究任务。而部分企业只想享受待遇,而不想尽到责任;就存在采用优先审评程序上市后不规范开展补充研究的情况。FDA将在2025年发布相关指南,督促企业完成。
※NDC Creation, Assignment, Listing and Appropriate Use for Human Drugs, Including Biological Products
翻译:人用药物(包括生物制品)的NDC创建、分配、登记和适当使用指南。
对于在美国市场流通的药品,都需要向FDA申请NDC号码;这是用于产品生命周期追溯的一个关键号码。但是提醒读者注意,这个号码是用于追溯的,不代表产品获得FDA审核或者批准。这个号码和国内批准文号不是一个概念。
◆Biosimilars专题
※Biosimilar and Interchangeable Biosimilar Products: Considerations for Container Closure Systems and Device Constituent Parts
翻译:生物类似药和可互换生物类似药产品:容器密封系统和器械组成部分的注意事项。
生物类似药和参比生物制品的可比性关系,远超过化学仿制药和化药参比制剂的关系。
为了证明生物类似药满足技术要求,可以在临床上替代参比生物制品,每个国家审评机构都设置了很多技术要求。
生物类似药要上市,需要和参比生物制品进行头对头对比。FDA发布这份新指南,将指引企业在研发生物类似药方面,关注包材的可比性。
◆Clinical/Medical专题
※Considerations for the Inclusion of Older Adults in Clinical Trials
翻译:将老年人纳入临床试验的考虑因素
FDA发布这份指南,将指导企业在受试者范围涵盖老年人时,需要考虑哪些影响因素;将促进临床试验的规范开展。
◆Generics专题
※180-Day Exclusivity: Questions and Answers
对于首仿药给与180天的行政保护,这项措施已经实施多年。为了更细致解释这项政策,FDA将出台配套指南。这项工作被列入2024年指南工作计划,未完成。FDA将在2025年工作计划中,继续推荐这份指南。
※Refuse-to-Receive
ANDA Submissions-Refuse-to-Receive for DMF Facilities Deficiencies
翻译:DMF中设施缺陷导致的ANDA申报的拒绝接受
ANDA Submissions-Refuse-to-Receive Standards: Questions and Answers
翻译:ANDA申报拒绝接受标准问答。
FDA为了提高ANDA申报效率和质量,发布了很多份RTR指南;这些指南为行业指明了方向,需要企业技术人员反复研读。
※ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin
翻译:仿制rDNA来源参比制剂的某些高纯度合成肽药品的ANDA。
中国一致性评价工作正如火如荼的开展,目前一致性评价范围覆盖口服药品和注射剂2个大类。根据2025年初国务院的53号文,2025年中国将把一致性评价药品范围拓展到贴剂等范围。
而美国仿制药的范围已经拓展到rDNA来源参比制剂为被仿制对象的合成多肽类仿制药;因此不得不佩服FDA对药品研发和监管科学的推动工作。
※半固体制剂的技术指南
In Vitro Permeation Tests for Semisolid Topical Products Submitted in ANDAs
翻译:在ANDA中提交的半固体局部产品的体外渗透测试指南。
In Vitro Release Tests for Semisolid Topical Products Submitted in ANDAs
翻译:在ANDA中提交的半固体外用产品的体外放行测试指南。
半固体制剂属于特殊剂型,对于这类产品的研发和注册,FDA已经发布了很多PSG来指导。在2025年,FDA将发布2份指南,阐释类似问题。
◆ICH专题
※M4Q(R2) Addressing Common Technical Document (CTD) Quality-Related Questions
翻译:ICH M4Q(R2) 解决通用技术文档 (CTD) 质量相关部分的问题
ICH M4已经发布多年,为国际药品注册和贸易发挥了重大作用。然而,现行版CTD-Q部分确实太陈旧了,从格式看主要适用于化药和重组类生物制品,不适用于近几年研发热点,例如细胞基因治疗产品、药品-器械组合产品等。这次修订,将对类似问题进行阐释。
※Q1/Q5C Targeted Revisions of ICH Stability Guidelines
翻译:ICH Q1/Q5C-稳定性试验指南修订
这份指南很重要,是药品研发和注册阶段非常重要的内容。然而,由于过去国内对这些工作不重视,导致国内企业很多人员对于类似技术信息不关注。根据网络流传信息,ICH将把ICH Q1系列指南修订为核心指南+配套附录的组织形式。
◆Pharmaceutical Quality CGMP
※Responding to Form FDA 483 Observations at the Conclusion of a Drug CGMP
翻译:对药品CGMP结论的FDA 483表格观察项的回应指南
FDA在对各类企业检查后,如果有缺陷,会签发一份表格,就是行业内都关注的483表格。如果企业回复483所列缺陷不能令FDA满意,或者回复时间较晚,就会被FDA警告。过去FDA针对如何回复483无具体要求,因此行业内人士对此问题很困惑。这份指南编制工作在2024年已经被列为工作计划之一,没有完成,这次仍然被列为2025年工作计划之中。
◆Pharmaceutical Quality/CMC
※Container Closure Systems for Drugs, Including Biological Products
翻译:药品和生物制品容器密封系统指南
这份指南上一版本还是1999版,到现在已经26年了。虽然从内容看,这份1999年指南还算齐全,但是毕竟行业发生了巨变,这份老指南需要被修订。这份指南编制工作在2024年已经被列为工作计划之一,没有完成,这次仍然被列为2025年工作计划之中。
扫码领取CPHI & PMEC China 2025展会门票
【智药研习社近期直播】
来源:CPHI制药在线
声明:本文仅代表作者观点,并不代表制药在线立场。本网站内容仅出于传递更多信息之目的。如需转载,请务必注明文章来源和作者。
投稿邮箱:Kelly.Xiao@imsinoexpo.com
▼更多制药资讯,请关注CPHI制药在线▼
点击阅读原文,进入智药研习社~
基因疗法细胞疗法优先审批申请上市
100 项与 MidAmerica Health, Inc. 相关的药物交易
登录后查看更多信息
100 项与 MidAmerica Health, Inc. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月25日管线快照
无数据报导
登录后保持更新
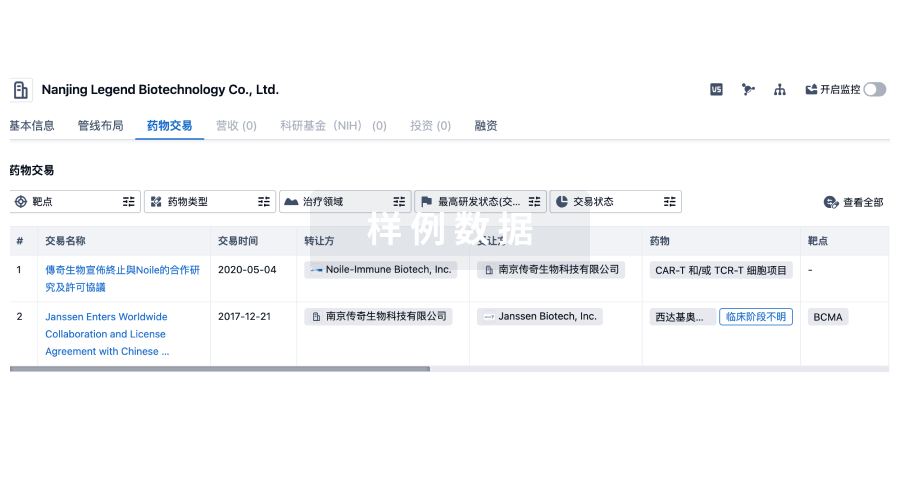
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
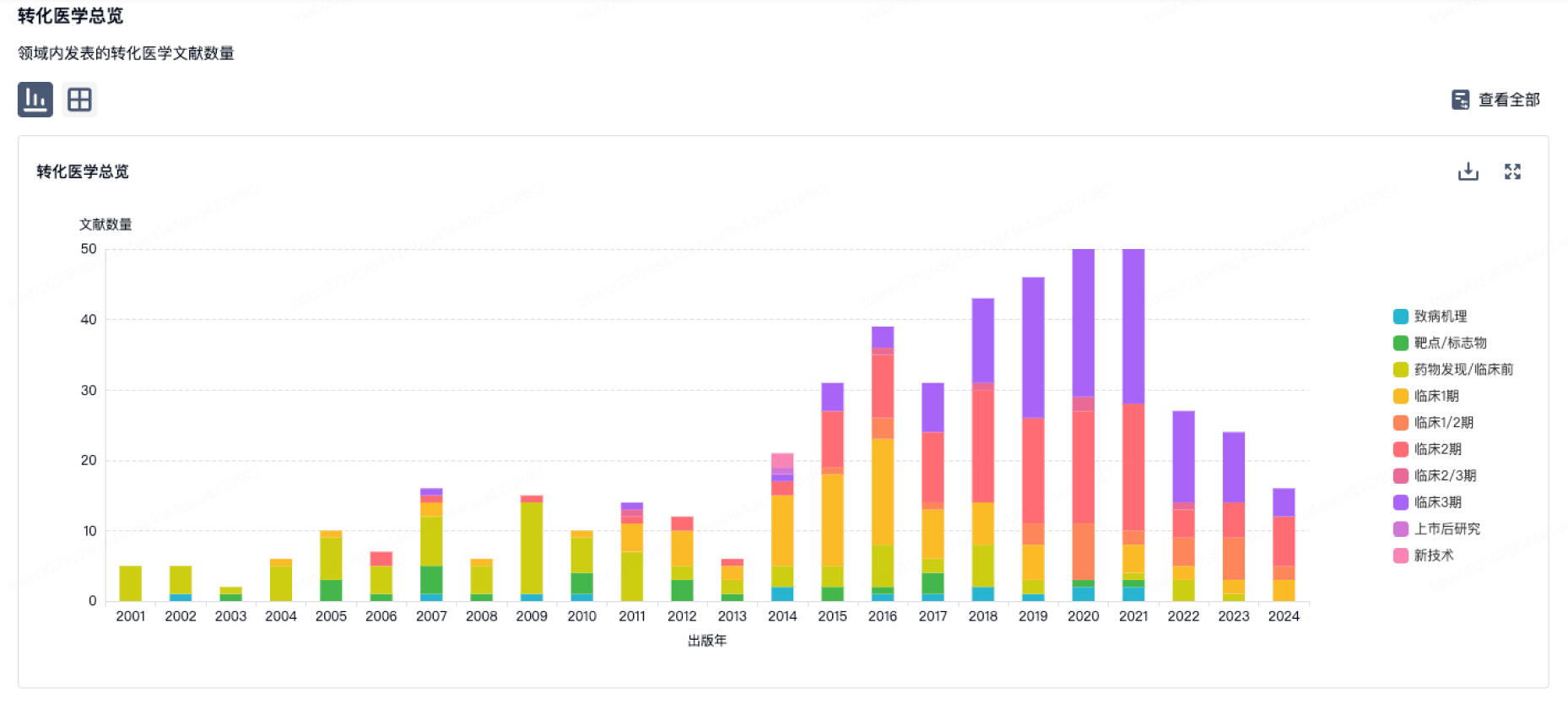
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
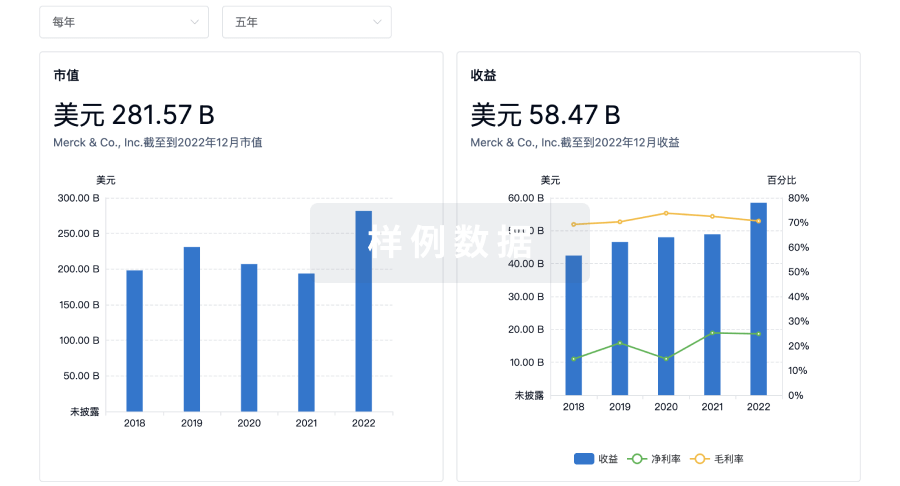
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
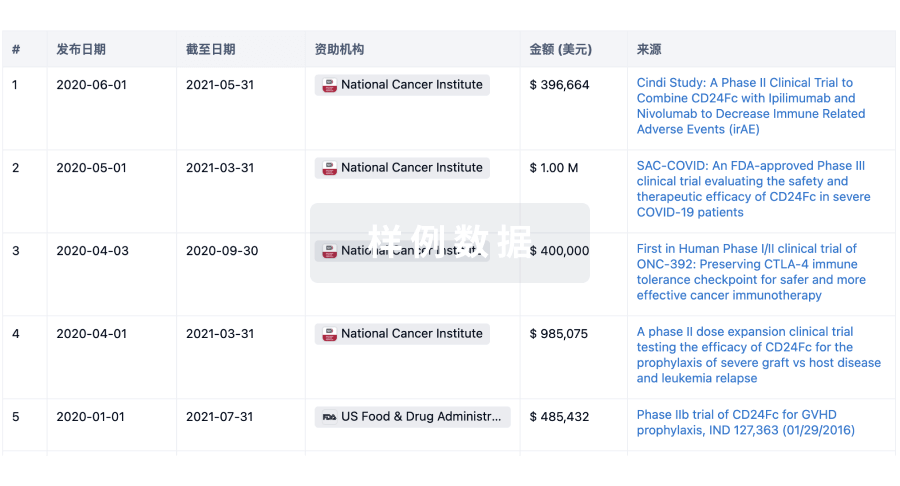
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用