预约演示
更新于:2026-06-14
Nanjing Vazyme Biotech Co., Ltd.
更新于:2026-06-14
概览
标签
神经系统疾病
细胞疗法
疾病领域得分
一眼洞穿机构专注的疾病领域
技术平台
公司药物应用最多的技术
靶点
公司最常开发的靶点

暂无数据
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
100 项与 南京诺唯赞生物科技股份有限公司 相关的临床结果
登录后查看更多信息
登录后查看更多信息
2025-12-01Alzheimers & Dementia
Apolipoprotein E ε4 homozygosity exacerbates retinal and cerebral microvascular dysfunction in Alzheimer's disease: A mediation analysis of vascular contributions to cognitive decline
Article
作者: Youjie Wang ; Yinhe Liu ; Hanyu Zhu ; Jiajing Qian ; Bo Tang ; Zhen Wang ; Jianing Shen ; Ho Ko ; Xiaoqian Luan ; William Robert Kwapong ; Lin Cao ; Yuntao Liu ; Caiyun Wen ; Jiahui Chen
Abstract:
INTRODUCTION:
Apolipoprotein E (APOE) ε4 is the strongest genetic risk factor for Alzheimer's disease (AD), with homozygous carriers (ε4/ε4) experiencing accelerated cognitive decline. While its role in amyloid and tau pathology is established, its impact on retinal and cerebral microvasculature remains underexplored.
METHODS:
A total of 107 AD (46 non‐carriers, 42 heterozygotes, 19 homozygotes) underwent optical coherence tomography angiography (OCTA) to assess retinal microvasculature and magnetic resonance imaging (MRI) ‐derived peak width of skeletonized mean diffusivity (PSMD) to evaluate cerebral small vessel disease. Plasma biomarkers (Aβ
42
, p‐tau217, glial fibrillary acidic protein [GFAP], neurofilament light chain [NfL]) and cognitive scores were also analyzed.
RESULTS:
Homozygous APOE ε4 carriers exhibited the most severe reduction in retinal microvascular density and higher PSMD (
p
< 0.001). Superficial retinal vessels and PSMD partially mediated APOE ε4's association with cognitive impairment.
DISCUSSION:
APOE ε4 homozygosity exacerbates retinal and cerebral microvascular dysfunction, which partially mediates cognitive impairment in AD.
Highlights:
Apolipoprotein E (APOE) ε4 homozygosity is associated with the most severe reductions in retinal microvascular densities and elevated cerebral small vessel disease (peak width of skeletonized mean diffusivity [PSMD]) in Alzheimer's disease (AD).
Vascular dysfunction (retinal and cerebral) correlates with lower Aβ42, higher p‐tau217/Aβ
42
, and worse cognitive scores (Mini‐Mental State Examination [MMSE], Montreal Cognitive Assessment [MoCA]).
Mediation analysis reveals that retinal (superficial vascular complex [SVC]) and cerebral (PSMD) microvascular changes partially explain the link between APOE ε4 and cognitive decline.Findings highlight vascular pathways as potential therapeutic targets in APOE ε4 carriers to mitigate cognitive impairment.
2025-04-01VETERINARY MICROBIOLOGY
Constructions and immunogenicity evaluations of two porcine epdemic diarrhea virus-like particle vaccines
Article
作者: Sun, Min ; Yang, Shanshan ; Xu, Hong ; Song, Xu ; Qian, Jiali ; Yang, Mengdi ; Zhou, Jinzhu ; Guo, Rongli ; Li, YuPeng ; Zheng, Fangyuan ; Li, Bin ; Wang, Chuanhong ; Zhao, Yongxiang ; Li, Yunchuan ; Liu, Shiyu ; Fan, Baochao
Porcine epidemic diarrhea virus (PEDV), a swine enteropathogenic coronavirus, causing acute diarrhea, dehydration, and up to 100 % mortality in neonatal suckling piglets, leading to huge economic losses in the global swine industry. Vaccination remains the most promising and effective way to prevent and control PEDV. In this study, we produced PEDV virus-like particles (VLPs) composed of S, M, and E proteins with a baculovirus expression system and a mammalian expression system. The S, M, and E proteins were effectively expressed and successfully assembled into VLPs. Subsequently, S subunits and commercially inactivated vaccines were selected and compared with two VLPs vaccines for immune efficacy through mouse immunization. The results showed that both VLPs induced higher levels of IgG, IgA, and neutralizing antibody titers, lymphocyte proliferation indexes and T, B cell ratios. Compared with the baculovirus VLPs, the mammalian VLPs exhibited better effects in inducing neutralizing antibodies, lymphocyte proliferations, and IFN-γ. These data indicated that the PEDV VLPs vaccine constructed using the mammalian expression system has better immune efficacy and has the potential to serve as a novel PEDV vaccine.
2025-02-01CCS Chemistry
Overcoming Cancer Chemoresistance Through Chromatin Compaction with Orally Platinum-Based ACLY Inhibitors
作者: Wang, Xiaoyu ; Nie, Junwei ; Zou, Lina ; Guo, Zijian ; Zhang, Shuren ; Wang, Weiqing ; Song, Dongfan
Chromatin remodeling critically influences gene expression and contributes to chemotherapy resistance in cancer.We introduce two Pt-based ATP citrate lyase (ACLY) inhibitors, PSN and PDN, which induce chromatin compaction to counter cisplatin (CDDP) resistance.PDN displayed significant cytotoxicity against CDDP-resistant cancer cells and demonstrated potent in vivo anticancer activity upon oral dosing with minimal toxicity.Mechanistically, PDN efficiently entered cancer cells, inducing DNA damage, downregulating ACLY, inhibiting DNA damage-induced histone acetylation, promoting chromatin compaction, and consequently suppressing genes linked to CDDP resistance.PDN, an oral metal-based ACLY inhibitor, represents a pioneering approach in conquering chemoresistance via chromatin remodeling, offering new insights for designing next-generation anticancer metallodrugs.
100 项与 南京诺唯赞生物科技股份有限公司 相关的药物交易
登录后查看更多信息
100 项与 南京诺唯赞生物科技股份有限公司 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月17日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
药物发现
1
登录后查看更多信息
当前项目
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
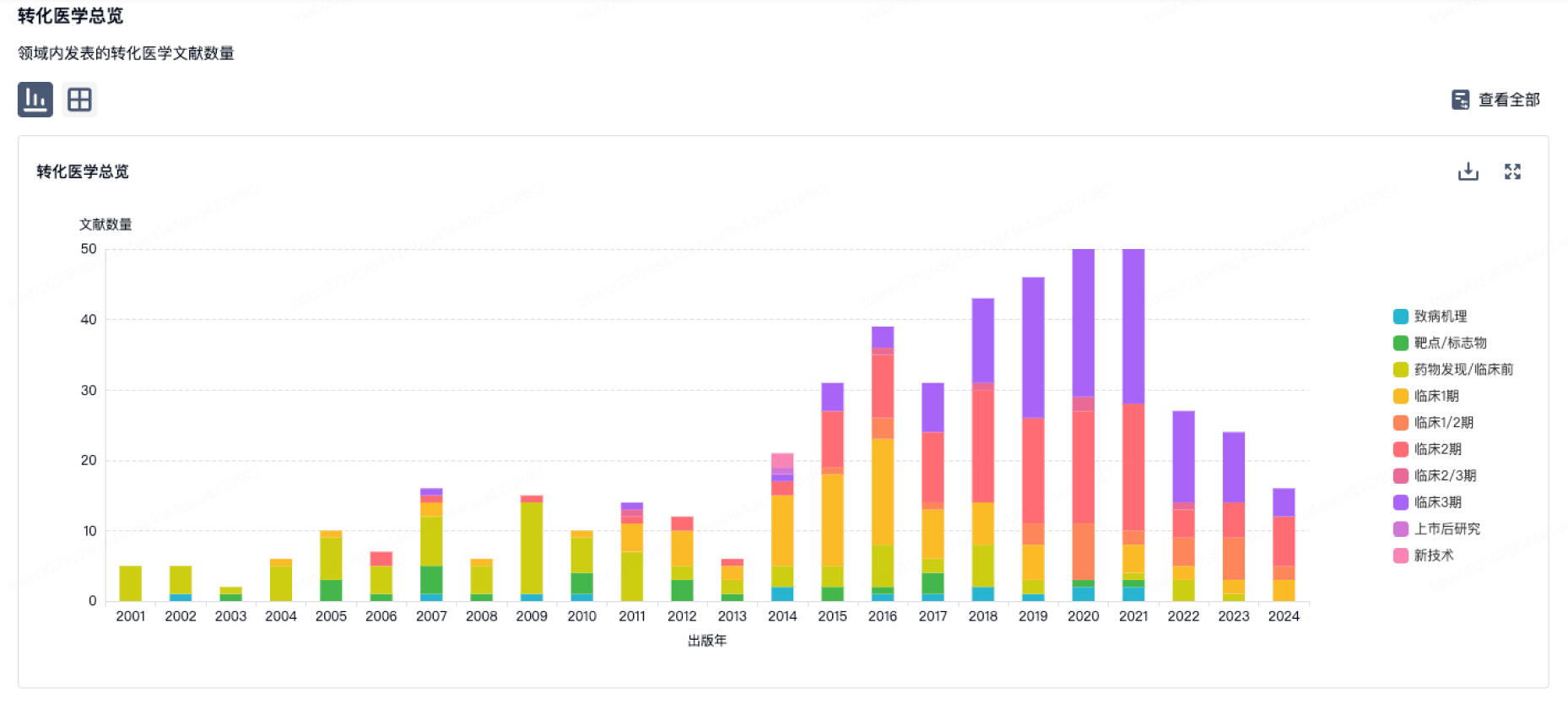
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
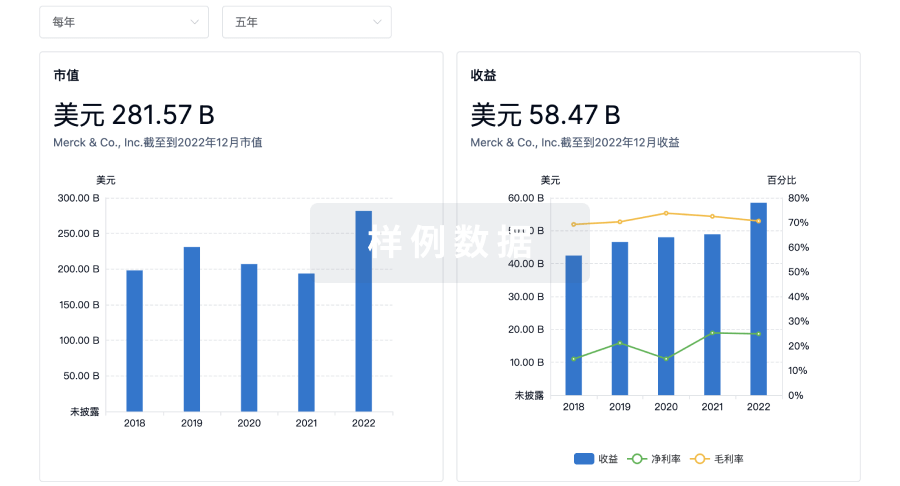
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
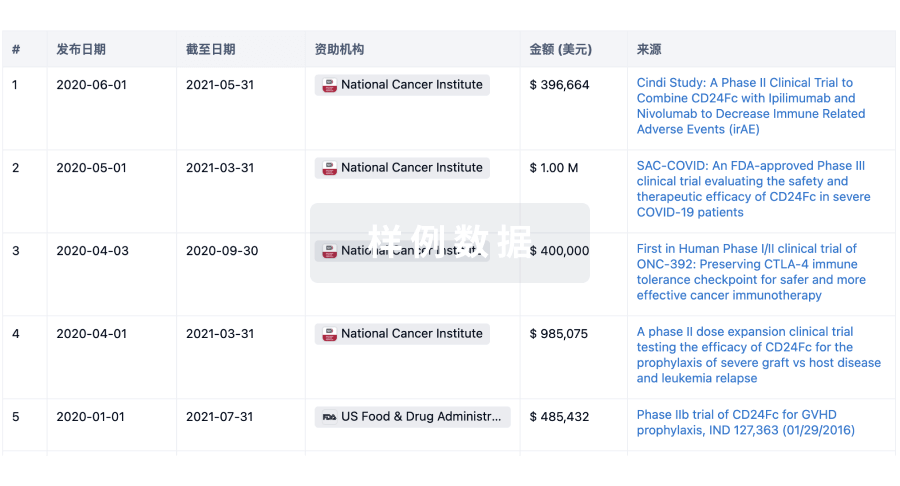
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用