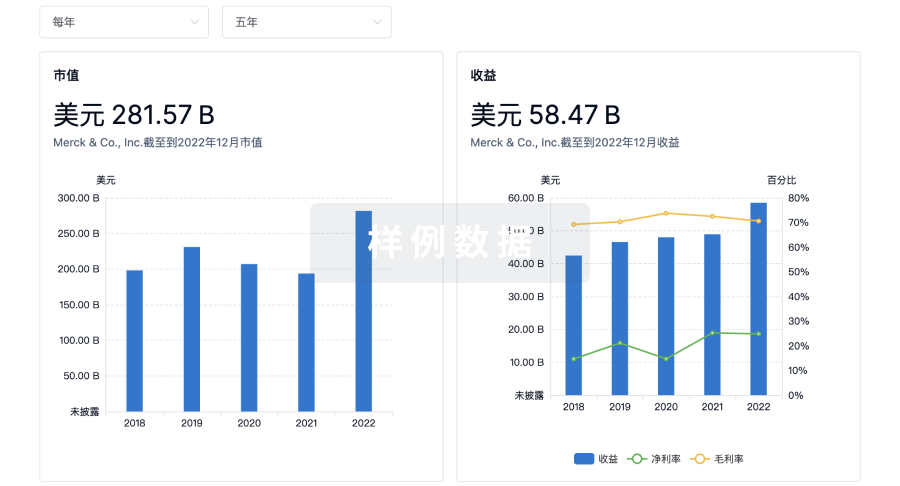
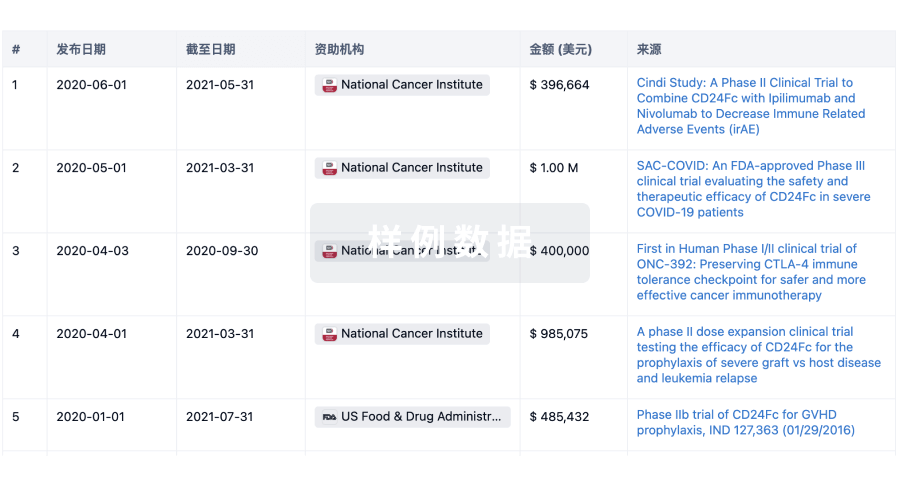
100 项与 上海泰楚生物技术有限公司 相关的临床结果
0 项与 上海泰楚生物技术有限公司 相关的专利(医药)
点击蓝字 关注我们
Legal express
【法规快讯】第9期生物制品GMP相关法规指南汇总(2026年3月-4月)
本文件是由泰澧生物汇总的中国药品监督管理局、US FDA\EDQM\USP等在2026年3月1日至4月30日期间发布的中外生物制品相关法规、指导原则以及其他相关重要信息,希望能为您的工作提供帮助。
01
中国国家药品监督管理局NMPA
国家药监局综合司关于做好生物制品分段生产有关工作的通知
发布日期:2026.04.02
内容简介:生物制品分段生产试点工作开展以来,国家药监局统筹推进,取得阶段性成效。新修订《药品管理法实施条例》(以下简称《条例》)对药品分段生产作出明确规定,即将正式施行。为加强政策衔接,保持政策连续性、稳定性,现就生物制品分段生产有关工作通知。
网页地址:https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20260402100454129.html
02
中国国家药品监督管理局药品审评中心CDE
关于公开征求《治疗用重组蛋白药物上市许可申请质量综述资料撰写指导原则(征求意见稿)》等三个指导原则意见的通知
发布时间:2026.03.09
内容简介:为进一步规范并提高相关产品上市许可申请的药学研究资料质量,基于ICH M4Q(R2)总体框架,结合各类产品的特点,我中心起草了《治疗用重组蛋白药物上市许可申请质量综述资料撰写指导原则(征求意见稿)》、《生物类似药上市许可申请质量综述资料撰写指导原则(征求意见稿)》、《抗体偶联药物上市许可申请质量综述资料撰写指导原则(征求意见稿)》。
网页地址:
https://www.cde.org.cn/main/news/viewInfoCommon/0fe53659e7c18d4cb81172309f83eb95
03
国家药品监督管理局食品药品审核查验中心CFDI
关于发布《制药用水检查指南》的通告
发布时间:2026.03.30
内容简介:为进一步加强检查员检查能力,指导药品生产检查员开展制药用水现场检查,核查中心组织制定了《制药用水检查指南》。
网页地址:
https://www.cfdi.org.cn/cfdi/resource/news/16700.html
以上资料信息来源:中国国家药品监督管理局、中国国家药品监督管理局药品审评中心、中国国家药典委员会、国家药品监督管理局食品药品审核查验中心、U.S. Food and Drug Administration、European Directorate for the Quality of Medicines、ICHhttps://www.usp.org/官方网站
关于泰澧生物
泰澧生物作为泰楚集团(CRDMO)旗下子公司,主要提供大分子生物药CDMO服务。在苏州工业园区拥有GMP生产厂房设施,面积约8000多平方米。满产计划每年70批次以上。泰澧生物的服务定位于:细胞系构建,工艺和分析方法开发及转移,GMP原液生产,制剂灌装,临床IND申报和上市BLA申报支持等。泰澧生物的生产设施和质量体系具有四年以上的成熟经验。成功支持5个中美澳IND项目申报,团队具有丰富的双抗药工艺开发和生产经验。泰澧生物专注于为生物技术公司和生物制药企业提供:稳定的质量,灵活快速的方案和交付。
咨询请联系:Technical_Support@tarlead.com
点击蓝字 关注我们
2025年6月3日13时许
北京大兴区一居民小区内
一辆停在垃圾分类处的货车突然起火
让人意想不到的是
引火“元凶”为三桶水
“不好,起火了,快来灭火!”
随着一声大喊,社区巡逻的微型消防站人员奔向着火的货车,使用灭火器先行处置,火势第一时间被成功控制后扑灭。
△为防止发生复燃,附近消防站消防员到达现场后,对现场进行进一步清理。
监控视频还原火灾经过
事后,火灾调查人员开展原因调查,在查看起火周边监控视频时发现,引发此次火灾的“元凶”竟然是放在货车顶部的桶装水。
监控视频显示,事发前货车车头顶上放置了三桶桶装水,当日中午太阳直射,桶装水下方垫着的纸箱先是冒出缕缕白烟,随后烟雾越来越大,周边的可燃物发生燃烧。
消防部门最终认定,此次火灾为阳光照射桶装水后,折射形成聚焦,引燃周围可燃物所致。
桶装水为何会引火?
透明塑料包装的桶装水、瓶装水在太阳光的照射下,会产生“凸透镜聚光效应”。
△图源:中国消防
在消防模拟试验中,光线经过瓶装水折射聚焦后,照射面上的温度可达120℃以上,若此时周边有易燃可燃物,易引发火灾事故。
山东航空学院理学院物理学博士刘慧解释,瓶装水通常是圆柱形,结构符合凸透镜特征。光线进入瓶装水内部介质时会改变传播方向发生折射。光源发出来的光经过凸透镜,会向中心位置汇聚,能量也随之汇聚,从而使温度升高。
除了瓶装水外,眼镜、水晶球等在阳光的照射下也会产生凸透镜聚光效应。
如何避免阳光聚焦引发火灾?
放大镜、矿泉水瓶、水晶球、眼镜等物品尽量避免放置在家中、车内阳光直射的位置;
这些物品尽量不要与可燃物毗邻放置;
夏季高温炙烤下,汽车内部温度会迅速飙升,下车时一定要随手带离此类物品,切忌遗留在车内;
此外,家长要加强对家中儿童的教育,不要因好奇而使用放大镜等聚焦光线长时间照射家中物品,谨防引发火灾。
夏季烈日将至
请务必检查家中、车内
放大镜、瓶装水、玻璃制品等
透光反光物品的摆放位置
避免阳光直射引发聚焦火灾
平安迎接夏日 从细节做起
图文来源:国家应急广播、央视新闻、内江消防。如有侵权请及时联系我们,我们将第一时间处理。
关于泰澧生物
泰澧生物作为泰楚集团(CRDMO)旗下子公司,主要提供大分子生物药CDMO服务。在苏州工业园区拥有GMP生产厂房设施,面积约8000多平方米。满产计划每年70批次以上。泰澧生物的服务定位于:细胞系构建,工艺和分析方法开发及转移,GMP原液生产,制剂灌装,临床IND申报和上市BLA申报支持等。泰澧生物的生产设施和质量体系具有四年以上的成熟经验。成功支持5个中美澳IND项目申报,团队具有丰富的双抗药工艺开发和生产经验。泰澧生物专注于为生物技术公司和生物制药企业提供:稳定的质量,灵活快速的方案和交付。
咨询请联系:Technical_Support@tarlead.com
扫码关注我们
崇德求真丨成就客户
点击蓝字 关注我们
(GB 46768-2025)《有限空间作业安全技术规范》
作为强制性国家标准,
于2025年10月31日发布,
2026年5月1日正式实施!
核心是构建“全流程闭环管控”体系,
坚守“先通风、再检测、后作业”底线,
适用于工贸、非煤矿山、建设等行业,化工、危化等特殊行业仍执行专用标准,
是防范有限空间作业安全事故的核心依据。
《有限空间作业安全技术规范》新版内容如何?
我们第一时间给大家分享。
↓ ↓ ↓
图文来源:新吴区建设工程安全质量监督站。如有侵权请及时联系我们,我们将第一时间处理。
关于泰澧生物
泰澧生物作为泰楚集团(CRDMO)旗下子公司,主要提供大分子生物药CDMO服务。在苏州工业园区拥有GMP生产厂房设施,面积约8000多平方米。满产计划每年70批次以上。泰澧生物的服务定位于:细胞系构建,工艺和分析方法开发及转移,GMP原液生产,制剂灌装,临床IND申报和上市BLA申报支持等。泰澧生物的生产设施和质量体系具有四年以上的成熟经验。成功支持5个中美澳IND项目申报,团队具有丰富的双抗药工艺开发和生产经验。泰澧生物专注于为生物技术公司和生物制药企业提供:稳定的质量,灵活快速的方案和交付。
咨询请联系:Technical_Support@tarlead.com
扫码关注我们
崇德求真丨成就客户
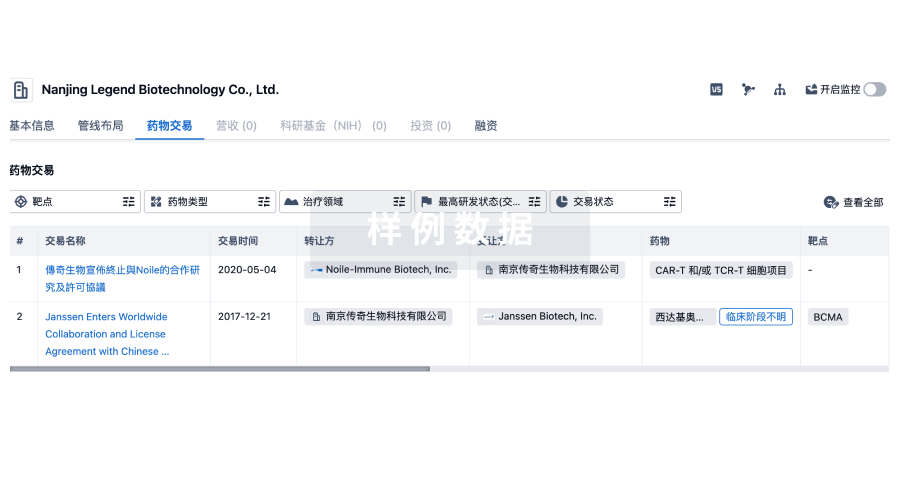
100 项与 上海泰楚生物技术有限公司 相关的药物交易
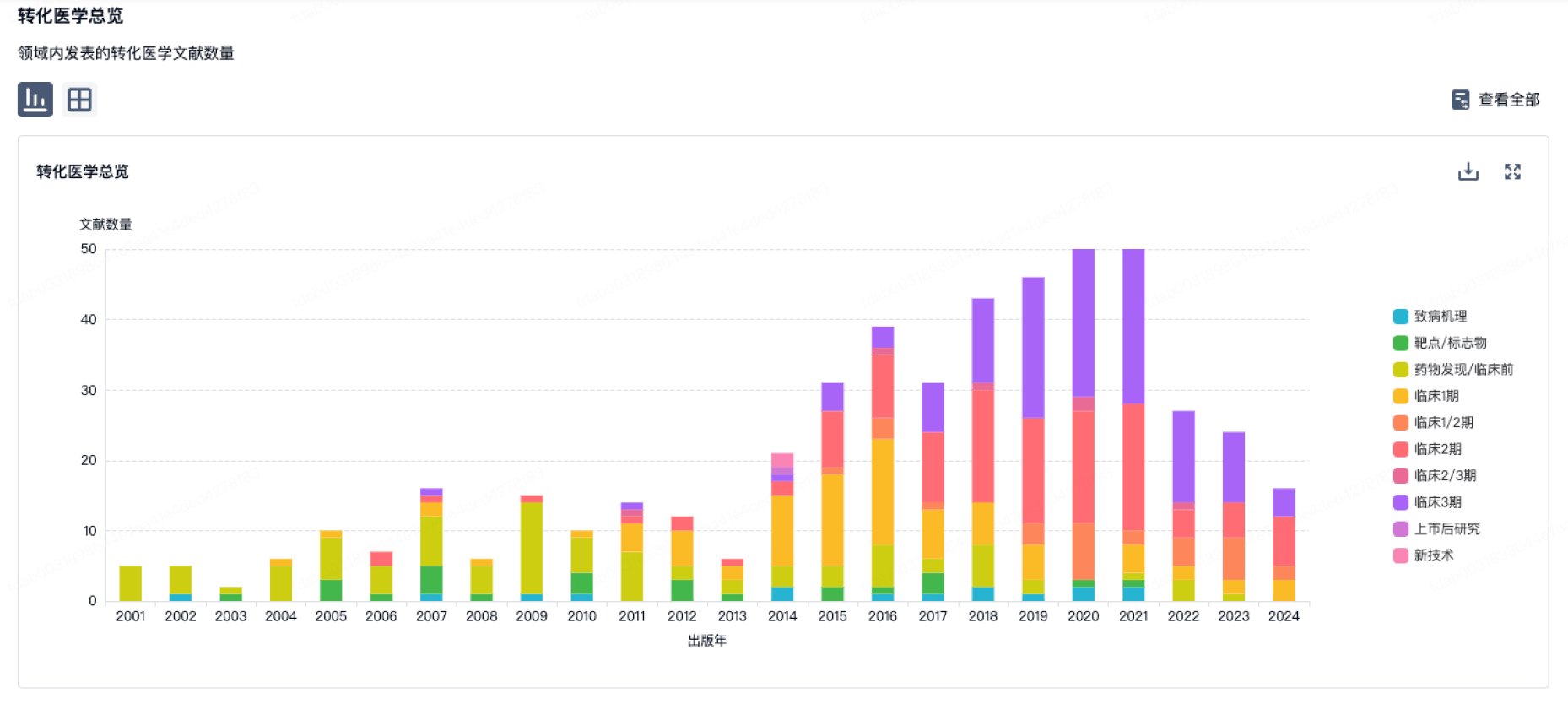
100 项与 上海泰楚生物技术有限公司 相关的转化医学