预约演示
更新于:2025-08-14

Nature Technology
更新于:2025-08-14
概览
关联
100 项与 Nature Technology 相关的临床结果
登录后查看更多信息
0 项与 Nature Technology 相关的专利(医药)
登录后查看更多信息
163
项与 Nature Technology 相关的新闻(医药)2025-08-13
2025年8月10日,一项在 RNA 治疗领域具有里程碑意义的研究成果重磅发布 ——北京大学/昌平实验室魏文胜教授团队与圆因生物携手,在国际知名期刊《Nature Communications》发表了题为 “Self-splicing RNA circularization facilitated by intact group I and II introns” 的研究论文。该研究提出了两种全新的体外RNA环化技术,不仅为RNA环化领域带来了革新,也凸显了圆因生物在环状RNA平台搭建方面的卓越实力。
— Nature Communications官网—
此次研究推出的两种技术分别是通过反式剪接进行的内含子-外显子置换(PIET)和用于RNA环化的完全自剪接内含子(CIRC)。其中,PIET借助I型内含子剪接的第二步,提供了一种独特的替代环化策略。而 CIRC 技术更是展现出显著优势,它利用I型和II型内含子的天然完整形式,无需进行内含子工程改造,这一特性使其在操作便捷性上远超传统方法。与内含子-外显子置换(PIE)相比,CIRC在温和条件下就表现出更高的 RNA 环化效率和更快的速度,为RNA 环化过程提供了高效保障。
利用CIRC技术,研究团队成功环化了编码全长肌营养不良蛋白(约12,206 个核苷酸)的 RNA,并实现了 427 KDa 蛋白的完整表达。这一重大突破,彻底打破了环化 RNA 编码蛋白大小的限制,为大分子量蛋白缺失相关疾病的治疗开辟了全新路径。
不仅如此,CIRC环化平台还具备诸多独特优势。通过 CIRC 技术能够生产无疤痕 circRNA 和免疫原性极低的 circRNA,这意味着其在临床应用中能有效降低不良反应的风险,提升治疗的安全性。同时,该平台可在内含子中引入 poly (A) 序列,从而与基于 oligo (dT) 的纯化体系兼容,极大地提高了 circRNA 生产和纯化的便利性,为其在研究和治疗应用中的规模化推进奠定了坚实基础。
图1: 通过PIET 和CIRC 进行RNA
北京:北京市海淀区永腾北路9号院18号楼
上海:中国(上海)自由贸易试验区李时珍路396号2幢1楼108-115室
寡核苷酸核酸药物
2025-07-11
·智药邦
自1999年首个基因组规模代谢模型(genome-scale metabolic model,GEM)问世以来,GEM已成为解析生物代谢的重要工具。该模型包含代谢基因、代谢物和反应,并结合化学计量矩阵与约束优化,系统描述和模拟生物体内的代谢过程。此外,GEM能够整合热力学参数、动力学参数、多组学数据及多细胞过程,构建更精细且具有更强大预测能力的多约束多过程模型。然而,先验知识的局限成为其发展的瓶颈。机器学习技术凭借强大的数据处理和模式识别能力,为进一步扩展GEM提供了新思路。近日,清华大学研究人员在《合成生物学》期刊上发表综述文章《机器学习驱动的基因组规模代谢模型构建与优化》。本综述系统总结了传统GEM及多约束多过程模型的构建流程,并着重探讨了机器学习在其中关键步骤中的应用前景,如基因功能注释、途径解析、空缺填补和生物学参数预测。机器学习技术作为新的驱动力,有望大幅度提升GEM的规模和质量,深化对生物代谢机制的理解,并推动实现数字孪生细胞。代谢过程是细胞内所有生物化学反应的集合,涉及能量的转化、物质的合成与分解以及细胞功能的维持。理解这些代谢过程对于阐明生物体的功能机制、诊断疾病、设计药物、设计细胞工厂以及优化生物制造过程至关重要。然而,由于代谢过程的复杂性,传统的实验方法在解析整个代谢网络时面临诸多挑战。为了克服这些困难,代谢模型应运而生。基因组规模代谢模型(genome-scale metabolic model,GEM)是一种整合基因、代谢物及反应的系统性框架,用于全面描述生物体内的代谢网络及其化学计量关系。该模型不仅能够借助优化算法预测代谢网络中的代谢流量,还可结合热力学参数、动力学参数、多组学数据及多细胞过程构建满足多样化研究需求的多约束多过程模型。自1999年发布首个流感嗜血杆菌的GEM以来,得益于大量完整基因组序列的测序和组装,目前已构建并发布了数千个物种的GEM。这些模型覆盖了细菌、酵母、植物和动物等多种生物,成为研究生物代谢网络的重要工具,广泛应用于细胞表型解析、代谢工程策略开发以及治疗靶点识别等领域。随着基因组测序技术和高通量组学技术的快速发展,GEM的性能和适用范围不断提升,这些进展极大促进了人们对生物体代谢机制的深入理解。然而,GEM依赖于现有科学知识的整合,其能力始终受限于当前已知信息。无论是代谢物、基因还是反应,即使是最先进的GEM,其知识覆盖范围仍存在不足,这在一定程度上限制了人们对生物代谢复杂性的全面认知。近年来,机器学习方法凭借其强大的模式识别与预测能力,已在大量学科得到广泛应用。尤其在生物学领域,机器学习技术已展现出显著优势,例如在蛋白质三维结构预测和抗体从头设计等领域,证明了其作为一种高效工具的潜力,能够基于已有知识深入探索未知空间。此外,该技术还在与GEM构建和应用相关的酶注释、途径挖掘和细胞工厂设计等方面展现出显著应用价值。本综述聚焦机器学习在GEM相关研究中的最新进展,重点探讨其在基因功能注释、代谢途径解析、代谢网络空缺填补,以及预测关键模型参数(如动力学、热力学和温度相关参数)中的应用。1 基于先验知识的代谢模型1.1 传统基因组规模代谢模型传统GEM的构建过程通常包括两个关键模块:粗略模型生成和模型修正(图1)。在粗略模型生成阶段,主要任务是对基因组中的代谢基因注释功能以及代谢反应的组装;在模型修正阶段,主要任务是整合遗漏途径与填补代谢网络中的空缺,此外还需要执行验证GPR(gene-protein-reaction)关系、估算生物量组成、整合转运和交换反应等任务。GEM的构建依赖于各种公共数据库,包括基因组数据库(如NCBI和KEGG)、蛋白质数据库(如UniProt、BRENDA、Expasy和SABIO-RK)以及代谢反应数据库(如KEGG、Rhea、MetaCyc和MetaNetX)。此外,一些数据库(如BiGG、BioModels和KBase)提供了已构建的GEM,可作为模型更新和新模型构建的模板。在模型构建完成后,必须经过一系列测试与评估,以确保其质量可靠性与形式规范性。图1 GEM以及多约束多过程模型的构建过程由于GEM的构建涉及大量数据的整合,Thiele等于2010年提出的GEM构建指南将GEM构建过程细化为96个步骤,但是这一过程烦琐且耗时。近年来新工具和自动化技术的发展显著加速了GEM构建过程。例如,COBRA Toolbox 3.0和RAVEN 2.0工具包提供了标准化平台,降低了模型构建和分析的学习成本。此外,ModelSEED、CarveME、Merlin、gapseq和AGORA2等自动化工具可完成模型构建的大部分步骤,包括基因组注释、GPR关系生成、反应可逆性和酶定位区室预测等。这些方法已广泛应用于细菌、古细菌和真核生物的模型构建,其中AGORA2为人类肠道菌群中的7302个菌株构建了GEM,展示了其在肠道微生物药物代谢预测中的潜力。尽管自动化工具和数据库的快速发展已显著提高了模型构建的效率,但基因功能的精准注释,尤其是对未知蛋白功能的表征,仍对模型的全面性和准确性构成挑战。因此,如何进一步克服这些限制,持续改进和完善GEM,依然是未来的关键任务。1.2 多约束多过程模型GEM主要涵盖代谢途径的信息,通常不包含其他代谢层次的约束信息。为克服这一局限,研究人员提出了更复杂的GEM,例如多约束模型、多过程模型(图1),以扩展代谢模型的应用范围。多约束模型通常不会增加基因数目,而是通过增加约束条件,使反应通量分布更加合理。与此不同,多过程模型在GEM的基础上进一步扩展,加入其他细胞过程,如蛋白质表达和分泌等,并将这些过程耦合起来,因此模型中基因、反应和代谢物数目会显著增加。凭借更大的规模和更高的预测精度,这些模型在工业菌株的理性设计中显示出巨大潜力。本文主要介绍不同类型多约束多过程模型及其构建所需的参数(图2),具体的模型原理已在其他相关综述中进行了详细讨论。图2 基因组规模代谢模型与多约束多过程模型的构建框架1.2.1 多约束模型现有的多约束模型主要包括热力学约束、酶约束、酶热约束以及酶温度约束等。其中热力学约束可以避免代谢通量计算中的不可行循环。热力学第二定律指出,正向反应的净通量对应于Gibbs自由能的负变化。热力学信息的整合可提升GEM的预测准确性,同时为异源代谢途径的筛选与评估提供依据。常用的热力学约束算法包括基于热力学的代谢通量分析(thermodynamics-based metabolic flux analysis,TMFA)、最大-最小驱动力(max-min driving force,MDF)分析以及OptMDFpathway。TMFA在常规通量平衡分析(flux balance analysis,FBA)中引入热力学约束,将可逆反应拆分为正向和反向,利用Gibbs自由能数据来评估反应可行性。MDF方法用于在满足代谢物浓度约束的解空间内,寻找可使途径中热力学瓶颈反应的驱动力最高的浓度分布。而OptMDFpathway框架进一步拓展了MDF方法的应用,利用混合整数线性规划(mixed-integer linear programming,MILP)在代谢网络中寻找热力学上可行的路径。酶约束模型(enzyme-constrained metabolic model,ecModel)是当前主流的多约束模型,其核心假设是细胞的蛋白质资源有限,需合理分配以确保各类生物过程的高效运转。ecModel通过在GEM中引入两大核心约束,进一步提高了代谢模型的性能。首先,反应通量受酶浓度限制,即每个代谢反应的速率受到参与该反应的酶浓度的限制。其次,细胞内总酶量有限,这意味着细胞必须在有限的蛋白质资源下合理分配,以确保不同的生物过程得以高效运行。这些约束使得模型能够更真实地反映细胞代谢过程,避免了传统GEM求解时出现的极端失真通量问题,从而在代谢工程和生物制造领域中,提供更加精确的预测结果。ecModel的框架包括FBAwMC、MOMENT及其简化版sMOMENT、ECMpy和GECKO等。细胞体积有限并对酶构成了空间限制。在此基础上,MOMENT和sMOMENT进一步扩展了模型的精度和适用性。MOMENT引入了更复杂的优化目标,首次使用蛋白质质量分数约束酶浓度。sMOMENT作为MOMENT的简化版,减少了计算复杂度,使得大规模代谢网络的分析更加高效。此外,2017年发布以来,GECKO框架因其代码公开和更高的自动化程度,且能够整合蛋白质组数据,已经广泛用于酿酒酵母(Saccharomyces cerevisiae)等多种生物的ecModel构建。ecModel的构建依赖于基因组模的酶活性参数。GECKO 3.0版本中引入了深度学习方法预测酶动力学参数,显著提升了模型的动力学参规模和预测精度。GECKO框架已帮助数百种生物自动化构建ecModel,推动了精准代谢分析的发展,在代谢工程、疾病模型、药物开发等多个领域取得了显著进展。除上述基础ecModel外,研究人员进一步结合其他约束条件扩展和优化模型,例如在酶约束基础上整合热力学约束或温度约束,适用于特定场景的代谢分析和表型预测。酶热约束模型依赖于酶活性参数和热力学参数,而酶与温度约束模型则通过整合蛋白质的最适温度(temperature optima,Topt)和熔解温度(melting temperature,Tm)计算酶在不同温度下的酶活性参数,调整对模型的约束。1.2.2 多过程模型多过程模型通过在GEM中纳入更多细胞过程,整合多个层次的细胞代谢信息,考虑细胞过程之间的相互影响,更精确地反映细胞的真实状态。此类模型将代谢物(如氨基酸、ATP和GTP)作为大分子(如酶、mRNA、tRNA和核糖体)的合成原料,而大分子的合成过程也会限制代谢反应速率,从而描述胞内资源的约束和分配。其中最常见的框架为代谢与表达(metabolism and expression,ME)模型和蛋白组约束模型(proteome constrained model,pcModel),两者的本质相同,主要是命名的差异。ME模型框架发展较早,目前已针对热厌氧单胞菌(Thermotoga maritima)、大肠杆菌(Escherichia coli)、隆德克氏梭菌(Clostridium ljungdahlii)以及枯草芽孢杆菌(Bacillus subtilis)等生物开发了ME模型。在基础模型框架外,研究者开发了FoldME、AcidifyME和OxidizeME框架,分别扩展了热、酸和氧化应激细胞过程,并针对大肠杆菌开发了相应模型,描述了在不同胁迫下蛋白质的重新分配与保护机制。StressME框架进一步将3种细胞应激过程整合,针对大肠杆菌开发了相应模型,应用于模拟在多种环境胁迫下的生物学响应。而pcModel发展稍晚,目前针对乳酸乳球菌(Lactococcus lactis)、酿酒酵母分别开发了pcLactis和pcYeast模型。在pcModel的基础上,针对酿酒酵母构建了pcSecYeast以及CofactorYeast,这两种模型框架分别整合了蛋白分泌过程和金属辅因子,扩展了多过程模型的细胞过程覆盖度和模拟适用范围。此外,ETFL(expression and thermodynamics flux framework)模型框架,在蛋白质表达过程的基础上,整合了热力学约束,并采用MILP取代传统模型中使用的迭代线性规划(linear programming,LP),显著提升了模型的预测能力。目前,ETFL框架已被用于构建大肠杆菌和酿酒酵母的模型。相较于已覆盖上百个物种的ecModel,多过程模型的物种覆盖度较低。这一局限主要源于其较高的复杂性和对参数的依赖。一方面,多过程模型需要整合更多的细胞过程,其所需的参数和过程信息尚未被充分表征;另一方面,目前仍缺乏成熟的自动化建模框架,使得构建此类模型的难度进一步增加。具体来说,ME模型和pcModel的参数输入主要包括酶活性参数、酶复合体的组成以及各蛋白质组分的计量系数。而ETFL模型在此基础上引入了热力学约束,因此还需输入热力学参数。2 机器学习辅助传统基因组规模代谢模型构建尽管GEM在过去取得了显著进展,但仍面临许多挑战,这些问题限制了GEM的准确性和全面性。近年来,机器学习在基因注释、途径解析及空缺填补等方面发挥了重要作用,本文将从这些方面展开讨论(图3),表1汇总了其中与机器学习相关的方法及方法介绍。图3 机器学习辅助基因组规模代谢模型与多约束多过程模型构建表1 机器学习辅助基因组规模代谢模型的方法扩展2.1 机器学习辅助基因功能注释GEM的构建依赖于基因功能注释数据,许多未知功能的基因限制了GEM中基因和反应的规模。传统的基因功能预测方法通常依赖于在大型注释数据库中识别相似或同源序列(如BLASTp和HMMER)。在基因功能注释中,GO和EC编号被广泛应用。然而,由于GO无法直接关联具体的代谢反应,无法直接应用于代谢建模,因此本节重点讨论了基于EC编号的基因功能预测方法[图3(a)]。这一类方法通过训练机器学习或者深度学习模型预测目标蛋白的EC编号,相关的方法有EzyPred、SVM-prot、DEEPre、DETECT v2和ECPred。然而,这些早期方法面临着数据的局限。例如,DETECT v2和ECPred使用的数据集分别仅涵盖786和858种EC编号。ExPASy目前已经收录超过8000个EC编号,以上方法难以胜任基因组规模的基因功能注释任务,因此这些方法未在表1中进行总结。随着数据的日益丰富,DeepEC应运而生。DeepEC利用了来自UniProt的数据集,其中包含4669种EC编号,采用多任务模型架构,兼顾酶与非酶的分类预测以及EC编号的预测,在未出现在训练数据中的通过文献搜集的201个酶序列集合上预测精度为0.92,召回率为0.45。人工智能技术的不断发展,包括蛋白质大语言模型ESM系列模型和蛋白质三维结构预测模型的出现,也使得基因功能注释模型的性能得到了进一步的提升。CLEAN采用蛋白质的预训练模型ESM-1b生成嵌入向量表示,并引入对比学习框架,通过计算查询序列与每个EC编号嵌入向量的欧氏距离预测,有效缓解了EC编号分布不均的问题,能够高质量地注释研究较少的酶、纠正错误标记的酶以及识别具有两个或更多EC号的功能混杂酶。该方法成功注释了36种此前未注释的卤化酶,并通过体外实验验证了其高准确性。随后发表的DeepECtransformer,作为DeepEC的升级版,引入蛋白质大语言模型ProtBert以强化蛋白质表征,并通过移除数据集中少于100条序列的EC编号条目缓解了极端类别数据稀缺的问题,成功预测并表征了大肠杆菌中3种酶的功能(YgfF、YciO和YjdM)。此后发表的多个模型(例如基于分层次双核多任务学习架构的ECRECer、基于证实深度学习的ECPICK、基于Transformer架构的EnzBert,以及基于残差神经网络的EnzymeNet)的性能也都优于同源比对方法或早期的机器学习方法,但这些方法在发布时没有与CLEAN以及DeepECtransformer进行基准测试。随着蛋白质结构预测能力的显著提升,GraphEC首次在EC编号预测任务中整合了蛋白质三维结构信息,通过ESMFold预测蛋白质结构,并结合ProtTrans生成的序列嵌入提取蛋白质特征。GraphEC通过引入酶的活性中心进行EC编号预测,在两个独立测试集上取得了现有方法中最高的召回率和AUC。但是,由于GraphEC数据集中没有非酶标签,因此与CLEAN存在相似的缺陷,可能为非酶的蛋白质序列分配EC编号,在使用此类方法辅助GEM构建时,需要区分假阳性结果。利用各类EC编号预测模型对生物基因进行预测,并通过在KEGG等生化反应数据库中查询EC编号对应的反应,即可实现基因功能注释,对GEM传统构建流程中的基因功能注释环节起到完善或替代作用。2.2 机器学习辅助途径解析构建高质量GEM的另一个瓶颈是其代谢网络中许多代谢物的合成或降解途径尚未被完全解析。这些代谢物难以与现有的代谢网络连接,限制了GEM在代谢物和反应规模上的扩展。这一问题主要源于两个方面:首先,酶底物的混杂性问题,即一类酶能够催化多种在化学结构上与其标准底物相似的反应物;其次,存在尚未被揭示的酶反应机制。酶的混杂性长期以来未引起充分重视。研究显示,大肠杆菌中约37%的酶对与其主要底物结构相似的底物表现出混杂活性。为扩展反应空间,基于模板的逆合成方法被广泛应用于代谢途径预测。这类方法依赖从生化反应数据库中构建的反应模板库,通过匹配目标产物与适当模板,推测可能的反应物并生成相关反应路径[图3(b)]。已发表的基于模板的逆合成方法包括PathPred、RetroPath 2.0、novoPathFinder、RetroPath RL、AiZynthFinder、ASKCOS和chemoenzymatic-ASKCOS,在代谢途径挖掘中展现出重要的应用价值。然而,这些方法常面临组合爆炸的问题。为应对此问题,在部分方法中引入了机器学习方法,优化反应模板的选择,有效限制组合爆炸范围,并显著提升路径预测的效率与准确性。例如ASKCOS使用了前馈神经网络(feedforward neural network,FNN)预测与目标分子最相关的反应模板,减少假阳性反应数目和计算成本,确保预测途径的可行性,以最大化实验成功的可能性,但是ASKCOS只包含化学合成的模板。chemoenzymatic-ASKCOS额外整合了酶反应的模板实现混合合成路径设计。RetroBioCat使用基于深度神经网络(deep neural networks,DNN)训练的SCScore方法,通过对每个分子的复杂度进行评分,指导逆合成搜索选择更简单的起始分子。RetroPath RL使用基于蒙特卡洛树搜索的强化学习方法来选择最佳途径,探索深度比RetroPath 2.0提升1倍以上。为了突破对已知反应模板的依赖,探索未知的反应机制,一些研究开发了无模板的反应预测方法,这些方法直接预测反应物或生成物[图3(b)]。一个典型的例子是Kreutter等将USPTO数据集作为一般化学知识的来源,并将其迁移至从文献中收集的数千条酶催化反应数据,训练了一个基于反应文本表示的序列到序列预测模型,该模型通过将输入的反应物SMILES转换为生成物SMILES,实现对反应产物的预测,但是这个方法只考虑了正向预测的情况。Probst等在SMILES作为输入的基础上,整合了反应EC编号的输入来提升模型性能,还实现了逆合成的拓展。Zheng等基于Transformer架构开发的BioNavi-NP模型在有机反应数据和酶反应数据上训练,成功预测了多种天然产物的合成路径。Zeng等基于BioNavi-NP提出了改进版的BioNavi,通过在深度学习模型中引入多任务学习和反应模板,以更为直观和可解释的方式设计混合合成路径。无模板的方法为探索未知反应机制提供了新的路径,有效推动代谢网络和酶催化反应的扩展。这些方法都为挖掘未知反应提供了有效工具,可以进一步拓展反应空间,提升模型的覆盖范围与准确性,为构建完善的GEM奠定基础。途径解析过程中,新预测的生化反应尚未分配EC编号,无法通过现有EC编号预测模型进行酶注释,这使得将这些反应与特定基因对应成为挑战,进而影响了GEM中GPR关系的准确性。这一任务不仅对于天然代谢途径的酶挖掘至关重要,对于开发新型非天然代谢途径同样具有重要意义。目前,新反应的酶注释通常基于序列同源性及反应相似性等特征。例如,Selenzyme、EC-BLAST、RxnSim以及SelenzymeRF都是通过计算目标反应与已知酶注释的反应之间的相似性识别可能的酶催化反应,其中,Selenzyme通过基于参与反应的所有化合物的完整结构来计算反应相似性,而EC-BLAST、RxnSim则通过反应的分子指纹来计算。SelenzymeRF作为Selenzyme的更新版本,通过引入sim_RF算法,并结合RXNMapper实现反应中的原子映射标注进一步优化了推荐酶的能力。然而,需要指出的是,序列相似性与酶的功能相似性并不总是严格对应,因而仅依赖序列相似性有时不足以准确预测酶的催化能力。在这一背景下,涌现了许多酶-底物结合预测和酶-反应催化预测的深度学习模型来辅助酶挖掘[图3(b)]。传统的酶-底物结合预测方法依赖分子动力学模拟,尽管其精确性较高,但计算量庞大且耗时。然而,随着合成生物学和计算生物学的快速进展,酶反应的实验数据逐渐积累,机器学习方法逐步成为一种高效的替代方案。ESP模型采用ESM-1b和图神经网络(graph neural network,GNN)分别表征蛋白质序列和小分子,再结合这两种特征表示,在约18 000对经过实验验证的酶-底物数据集上训练梯度提升决策树模型,用于酶-底物结合预测。另一个例子是EnzRanK,其依赖卷积神经网络(convolutional neural network,CNN)获取酶序列和底物的特征,并生成结合概率评分。基于此评分,模型能够排序候选酶,识别出在新型底物上可能具有活性的酶。在酶-底物结合预测任务中,正样本可以从反应数据库中轻易获取,但是负样本的获取具有不确定性。一方面,直接将酶与非已知底物组合会导致正负样本数据失衡;另一方面,随机抽取的酶-底物对可能由于底物混杂性而实际属于正样本。这会导致模型的预测出现偏差。因此,PU-EPP模型中提出了一种结合正样本和无标签学习的策略,以最大限度减少不准确负样本的影响。PU-EPP成功鉴定了15种对赭曲霉毒素A和玉米赤霉烯酮具有特异性的降解酶。最近,Qian等使用ESM-1b表征蛋白质序列、GNN表征小分子,结合DNN训练了MEI模型,在从文献中手动收集的两个测试集上表现优于ESP。并且在MEI的基础上,利用专业数据集分别针对CYD抑制剂与底物预测任务与塑料降解酶预测任务进行了微调,均能达到特定领域预测模型的先进水平。这些方法的结合有效提高了酶-底物结合预测的准确性和筛选效率,可以帮助确定酶在代谢途径中的功能,为GEM构建提供更准确的GPR关系。与此同时,Hu团队开发了深度学习模型SPEPP,可以预测酶-底物-产物三元组发生反应的可能性。其优势在于克服了传统方法中反应相似性计算的局限,更加侧重于底物与产物之间的关系,拓宽了潜在酶催化反应的发现范围。此外,Shi等开发了一个综合平台REME来辅助酶挖掘和评估。该平台通过整合原子映射、原子类型变化以及基于分子指纹的反应相似性计算,实现与非天然反应相似的已知反应的快速排序并获取候选酶。REME还参考了酶-底物结合模型和动力学参数预测模型的预测结果,有助于在筛选和评估过程中迅速识别出候选酶。2.3 机器学习辅助模型空缺填补除了基因功能注释和途径解析,GEM修正过程中的空缺填补环节也受益于机器学习的快速发展[图3(c)]。GEM是基于目标生物体中所有已知的代谢反应和基因构建的。然而,由于对生物体的认识不完善,这些网络重建中常常存在空缺(gap)。为解决这一问题,Thiele等开发了fastGapFill算法,这是第一个能够高效检测并填补GEM网络缺口(gap-filling)的工具。该算法通过从通用生化反应数据库中筛选最小反应集合,将其整合到GEM中,使原本的死端反应可以具有通量。此外,Kumar等开发了GapFill算法,此方法以GEM中无法生成的代谢物为目标,改变反应方向或从MetaCyc数据库添加反应,将这些代谢物与现有底物连接起来。上述基于网络拓扑结构的空缺填补算法依赖于候选反应数据库,并且最小化候选反应集合的优化方式缺乏相应理论依据。随着机器学习的发展,也出现了新的空缺填补方法。Oyetunde等提出的BoostGAPFILL利用基于矩阵分解的机器学习和整数最小二乘优化方法填补代谢网络中的缺失反应,为空缺填补开辟了新的途径。Chen等于2022年提出了CHESHIRE(CHEbyshev Spectral HyperlInk pREdictor),该算法通过引入超图和CNN的概念,基于代谢网络的拓扑特征预测GEM中的缺失反应,仅需输入GEM,即可生成候选反应的置信评分,有助于快速识别GEM中的关键缺失反应并增强表型预测。Huang等开发的DSHCNet进一步考虑代谢反应的异质性,并在表示GEM的超图的顶点表示中明确区分底物和产物,基于超图结构实现GEM的空缺填补。此外,针对宏基因组组装过程中基因组不完整性导致的GEM空缺问题,Boer等提出了一种名为DNNGIOR的深度神经网络引导的反应集合推测方法。该方法能够基于不完整的反应集合预测潜在缺失的反应,补全GEM,但是DNNGIOR的预测性能受到代谢反应在细菌群体中的出现频率以及查询基因组与训练基因组之间系统发育距离的显著影响。3 机器学习辅助多约束多过程模型构建多约束多过程模型通过整合多个层次的细胞过程及其相互作用,进一步提升了对复杂生物系统的描述能力。这类模型通常更加复杂,所需的参数显著增加,包括酶动力学、热力学等多种数据。然而,当前实验技术解析的生物学参数有限,难以全面覆盖模型所需数据,成为了多约束多过程模型快速发展的瓶颈。为应对这一问题,许多基于机器学习的研究致力于这些参数的预测,从而扩展代谢模型的规模和质量,提高模拟精度,使模拟结果更贴近细胞的真实状态[图3(d)]。表2汇总了其中与机器学习相关的方法及方法介绍。表2 机器学习辅助多约束多过程模型获取参数方法3.1 动力学参数预测酶动力学参数在解析细胞代谢机制、蛋白质组分配及生理多样性中具有关键作用。其中,kcat(催化常数)定义了酶促反应的最大化学转化速率,Km(米氏常数)反映了酶达到最大催化速率一半时所需的底物浓度,kcat/Km则衡量酶的整体催化效率。在酿酒酵母的ecModel模型中,仅有约5%的酶促反应能够在BRENDA和SABIO-RK数据库中找到完全匹配的kcat。为支持ecModel的进一步扩展,研究者开发了多种动力学参数预测方法,以扩大其覆盖范围。现有kcat预测方法包括Heckmann等开发的方法、DLKcat、TurNuP、DLTKcat和DeepEnzyme,Km预测方法包括Kroll等开发的方法、GraphKM和MLAGO,以及兼顾了kcat、Km预测的MPEK,兼顾了kcat、Km和kcat/Km预测的UniKP和EITLEM-Kinetics。一些方法已成功应用于GEM模型优化与合成生物学领域。例如,DLKcat是首个专注于大规模预测kcat的深度学习工具,成功预测了343种酵母和真菌的kcat,并辅助构建了相应的ecModel。作为kcat预测的关键模块,DLKcat已被整合到酶约束模型自动化构建流程GECKO 3.0和ECMpy 2.0中。TurNuP进一步推动了kcat值预测的发展,特别是在应对与训练集序列相似性较低的酶时表现出卓越的鲁棒性,并在基于ecModel的蛋白质组预测中展现出更高的准确性。此外,DLTKcat支持在指定温度下预测酶的kcat,而UniKP进一步扩展了功能,能够在指定温度和pH条件下预测kcat,满足实际应用中对环境参数的需求。UniKP和EITLEM-Kinetics在预测酶突变体的kcat方面同样表现优异,其中UniKP通过kcat和kcat/Km预测指导蛋白质突变,获得了高活性的酪氨酸解氨酶(TAL)。相比之下,Km的预测方法尚未开发出适合的下游应用。除酶约束模型之外,动力学模型中也需要kcat和Km参数,随着酶活性预测模型的发展,这些预测方法也会在未来发挥出重要作用。3.2 热力学参数预测热力学分析的核心在于精准预测反应的标准Gibbs自由能变化(∆rG′⊖)。截至2019年,酶促反应热力学数据库(TECRDB)中仅收录了约600个酶促反应的实验测量热力学数据。为弥补实验数据的不足,研究者开发了基于基团贡献(group contribution,GC)的方法,并将其应用于代谢模型的构建。然而,基团贡献方法存在一些内在局限性,包括:①专家定义的基团覆盖范围有限,导致某些代谢物无法被分解,进而无法估算其∆rG′⊖;②对于无基团变化的反应(如异构反应),∆rG′⊖被赋值为零,而实验数据表明其实际值非零。Alazmi等提出了一种基于化学指纹特征的机器学习算法——指纹贡献(fingerprint contribution,FC)方法,用于预测生化反应的∆rG′⊖。该方法以二维化学指纹表示化合物特征,主要分为两个步骤:首先,从大量二维指纹特征中系统筛选相关特征;其次,利用正则化回归方法构建最终的线性预测模型。此外,Wang等开发了dGPredictor工具,该工具能够考虑代谢物结构中的立体化学信息,显著提升反应的覆盖率。dGPredictor还可预测新反应的∆rG′⊖,并可以集成至代谢途径从头设计工具,以避免在设计过程中引入方向性不可行的反应步骤。3.3 温度相关参数预测蛋白质作为一种执行大多数催化功能且对温度变化高度敏感的生物大分子在细胞中起着至关重要的作用。然而,整合温度的GEM建模仍面临挑战,主要原因包括代谢的复杂性以及缺乏足够带有温度信息的蛋白质属性数据。为了更好地模拟和预测温度对细胞代谢的影响,尤其是蛋白质在不同温度下的行为,目前已开发多种方法来预测蛋白质与温度相关的参数,包括Tm和Topt。早期的Tm预测方法通常采用one-hot编码、氨基酸组成等简单特征来表征蛋白质,然后使用传统机器学习模型预测Tm。随着深度学习技术的发展,研究逐渐采用了更复杂的模型。例如,Jung等开发了基于Transformer的DeepSTABp算法,该算法结合了热蛋白组分析的实验条件、氨基酸序列和宿主的最适生长温度(optimal growth temperatures,OGT)来预测Tm。此外,Li等提出了模型DeepTM,利用图卷积神经网络(graph convolutional neural network,GCN)和自注意力网络,通过序列信息直接预测蛋白质的Tm。该方法的蛋白质表征结合了宿主的OGT、进化信息、理化特性(包括疏水性、体积、极化率、等电点等)等特征。此外,机器学习模型Tome能够基于蛋白质氨基酸序列预测Topt。预测的Topt进一步应用于贝叶斯基因组规模代谢模型(Bayesian-GEM)中,以模拟温度对酿酒酵母细胞代谢的影响。研究揭示,当培养条件超过最适温度时,导致酵母生长受限的最关键限速酶是鲨烯环氧化酶(ERG1)。通过用耐热酵母菌株中的同源酶替代ERG1,获得了生长能力超过野生型的耐热菌株。值得注意的是,在Tome的训练数据集中,Topt小于85 ℃的样本占比超过95%,导致其在预测高温稳定酶时的能力不足。为了解决问题,Gado等提出了TOMER模型。TOMER在Tome模型的数据集上进行了重新训练,并通过重采样和集成学习策略缓解了数据不平衡问题,使得在测试集中Topt大于85 ℃的数据预测R2分数从0.52提升到0.63。为了更好地捕捉蛋白质序列与温度之间的关系,Li等利用包含300万种酶的OGT数据集训练了DeepET模型,并通过微调DeepET进而预测Topt和Tm。DeepET的微调模型预测仅依赖蛋白质序列,消除了此前Topt预测方法对OGT参数的依赖。类似的基于蛋白质序列的Topt预测方法还包括Preoptem,该方法采用one-hot编码表示蛋白质序列,并结合CNN进行训练。Preoptem助力从海洋宏基因组中挖掘到一种新的嗜热几丁质酶,展示了其在实际应用中的潜力。挑战与展望机器学习在GEM中的应用正逐步展现出巨大的潜力,尤其是在扩展GEM规模和质量、提升GEM细胞表型预测精度等方面。然而,尽管机器学习为GEM提供了更为强大的工具,当前常用的评估标准在应用到生物学领域时仍面临一定的局限性。常见的机器学习模型评估标准,如AUC、准确率和独立数据集的使用,通常受到数据分布的影响,容易导致高估模型性能和未能准确反映模型泛化能力的问题。这种问题在处理基因组规模的预测时尤为突出,过于依赖机器学习领域的评估标准可能忽视机器学习模型的普适性和多样性,特别是在零样本或少样本的预测场景下,可能导致对模型性能的误判。因此,未来研究需要开发更加完善的评估框架,以准确反映机器学习模型在GEM中的实际应用价值。此外,当前机器学习方法的应用仍然局限于传统机理代谢模型的框架内。机器学习更多地扮演着辅助角色,例如辅助基因功能注释、途径解析和参数预测等,而未能突破传统机理代谢模型的限制。许多细胞过程,尤其是那些难以精确数学表达的过程,仍然是当前GEM发展的挑战。因此,要充分发挥机器学习的潜力,未来的研究需要开发机理模型与数据驱动方法的深度融合框架,推动更多细胞过程的数学解析,实现对更复杂生物学系统的精准描述。通过将机器学习的多种模块有机结合,GEM不仅能够整合多尺度的生物学约束,还能够推动白箱与黑箱模型的融合,从而提升GEM的规模和性能。随着机器学习算法的进步,AI驱动的GEM自动化建模将逐步成为现实,这将大大提高其精度、速度和可靠性,推动其进入AI时代,进一步推动数字孪生细胞的实现。然而,数字孪生细胞的发展要求跨学科的深度合作。随着合成生物学和计算机科学的迅速发展,模型框架的优化需要生物学、计算机技术、数据分析等多个领域的紧密协作。这种跨学科的知识融合不仅对技术和方法的进步至关重要,更对研究人员的综合能力提出了更高要求。未来的成功将依赖于这一多元化知识体系的构建和创新。总的来说,目前机器学习在GEM中的应用仍局限于现有的框架,未来的研究应突破这些限制,推动机理与数据驱动方法的协同发展,从而为数字孪生细胞和精准生物学研究带来新的突破。通讯作者及团队介绍李斐然,清华大学深圳国际研究生院助理教授、博士生导师。曾荣获2022年国家优秀自费留学生奖学金,入选2023年国家海外高层次青年人才项目,入选《麻省理工科技评论》2023年中国区35岁以下科技创新35人,并获得AI100青年先锋等多项荣誉与奖项。长期致力于开发新方法与新技术,用于构建和解析数字生命模型,揭示生命系统的内在机理,推动合成生物学与生物医药领域的研究进展。近五年以第一或通讯作者身份在Nature Catalysis、Nature Communications、Molecular Systems Biology、Nucleic Acids Research和PNAS等国际高水平期刊发表SCI论文二十余篇。任BioDesign Research青年编委,并担任Nature Biotechnology、Nature Communications、PNAS等期刊的审稿人。参考资料:U Ke, LUO Jiahao, LI Feiran. Applications of machine learning in the reconstruction and curation of genome-scale metabolic models[J]. Synthetic Biology Journal, 2025, 6(3): 566-584--------- End ---------感兴趣的读者,可以添加小邦微信加入读者实名讨论微信群。添加时请主动注明姓名-企业-职位/岗位或姓名-学校-职务/研究方向。
核酸药物
2025-07-09
·学术经纬
ICOS:增强免疫疗效的新靶点在慢性感染和肿瘤微环境中,CD8+ T细胞常常会进入“耗竭”状态,导致免疫功能下降。近期发表在Immunity杂志上的研究揭示了这一过程发生的新机制:持续表达的共刺激分子ICOS会限制T细胞的抗感染和抗肿瘤能力。研究发现,阻断ICOS信号可以增强耗竭前体T细胞(Tpex)的功能,促进其分化为更具杀伤力的效应T细胞,并显著提升抗PD-1疗法的效果。这一发现为改善慢性感染和肿瘤免疫治疗提供了新思路。在慢性病毒感染模型中,具有ICOS缺陷的Tpex细胞能够进一步产生更多存活时间长、细胞因子分泌能力强的效应样细胞。而阻断ICOS配体(ICOSL)能够扩大CD8+T细胞的数量,降低病毒载量。在肝癌小鼠模型中,抑制ICOS同样增强了肿瘤特异性CD8+T细胞反应,改善了抗PD-1疗法对肿瘤的控制效果。这些发现表明,ICOS信号通路可能是优化现有免疫治疗策略的重要靶点。论文标题:The costimulatory molecule ICOS limits memory-like properties and function of exhausted PD-1+CD8+ T cellsDOI: 10.1016/j.immuni.2025.06.001研究开发新型单碱基编辑器据Nature Biotechnology的一项新研究,研究人员通过改造大肠杆菌tRNA特异性腺苷脱氨酶(TadA),开发出了能够实现单碱基精准编辑的新型工具。这项技术为基因治疗提供了更精确的工具,有望推动精准医疗发展。 传统碱基编辑器存在“广撒网”的问题,会修改编辑窗口内的所有同类碱基。而新方法通过定向进化技术,在核酸识别热点区域进行策略性筛选,最终获得了16种能够识别特定序列背景的脱氨酶变体。这些变体覆盖了目标胞嘧啶所有可能的上下游碱基组合,为定制化编辑器提供了丰富选择。 在实际应用中,这些新型编辑器展现出显著优势:在纠正ClinVar数据库中记录的疾病相关T:A到C:G突变时,81.5%的情况下比传统胞嘧啶碱基编辑器(CBEs)更准确。研究人员还成功在体外模拟了两种重要癌症驱动突变。这项研究为开发临床适用的精准基因编辑工具提供了通用策略,未来或将在遗传病治疗和癌症研究领域发挥重要作用。论文标题:High-precision cytosine base editors by evolving nucleic-acid-recognition hotspots in deaminaseDOI: 10.1038/s41587-025-02678-wCDK4/6抑制剂耐药机制CDK4/6抑制剂是治疗激素受体阳性、HER2阳性(HR+/HER2+)乳腺癌的重要药物,但耐药性问题一直困扰着临床治疗。近日,Nature Cancer发表的一项研究揭示了这一耐药现象背后的免疫学机制。研究人员发现,在CDK4/6抑制剂作用下,肿瘤微环境中的缺氧状态会激活CCL2依赖的信号通路,招募分泌白细胞介素-17A(IL-17A)的γδ T细胞浸润乳腺癌肿瘤。这些γδ T细胞促使肿瘤相关巨噬细胞(TAMs)向与抗药性相关的免疫抑制性CX3CR1表型极化,最终导致耐药性的发生。该研究在多个临床队列中验证了这一发现。此外,在接受新辅助PD-1阻断和放疗的HR+/HER2+乳腺癌女性患者中,CX3CR1 TAMs对预后有负面影响。这项研究阐明了HR+/HER2+乳腺癌对CDK4/6抑制剂耐药的免疫学机制,同时为开发针对γδ T细胞和CX3CR1+巨噬细胞的联合治疗策略提供了理论依据,有望为克服临床耐药问题开辟新途径。论文标题:IL-17A-secreting γδ T cells promote resistance to CDK4/CDK6 inhibitors in HRHER2 breast cancer via CX3CR1 macrophagesDOI: 10.1038/s43018-025-01007-z. 让麻疹病毒“停工”的联合疗法麻疹病毒是一种高度传染性的RNA病毒,每年在全球造成数百万人感染。尽管疫苗已大幅降低其危害,但目前尚无获批的抗病毒药物。最近,科学家们将目光聚焦于病毒复制过程中的关键“机器——聚合酶复合体,它由大蛋白(L)和四聚体磷蛋白(P)组成 ,是一个关键的抗病毒靶标。在最新发表于Cell的一篇研究里,研究人员通过冷冻电镜解析了麻疹病毒聚合酶复合体的结构,并发现两种非核苷抑制剂 ERDRP-0519 和 AS-136A能将其锁定在失活状态,从而阻止病毒复制。基于这些研究结果,科学家推测ERDRP-0519可能对同样缺乏特效药的尼帕病毒有效。尼帕病毒也是一种高致病性病毒,目前尚无获批的抗病毒药物。后续实验证实了这一猜想:通过解析尼帕病毒聚合酶的结构,证实ERDRP-0519对其转录活性也存在抑制作用。由此,该研究为对抗这两种致命病毒开辟了新途径。论文标题:Structures of the measles virus polymerase complex with non-nucleoside inhibitors and mechanism of inhibitionDOI:10.1016/j.cell.2025.06.017欢迎转发到朋友圈,谢绝转载到其他平台。如有开设白名单需求,请在“学术经纬”公众号主页回复“转载”获取转载须知。其他合作需求,请联系wuxi_media@wuxiapptec.com。免责声明:本文仅作信息交流之目的,文中观点不代表药明康德立场,亦不代表药明康德支持或反对文中观点。本文也不是治疗方案推荐。如需获得治疗方案指导,请前往正规医院就诊。更多推荐点个“在看”再走吧~
免疫疗法临床研究
100 项与 Nature Technology 相关的药物交易
登录后查看更多信息
100 项与 Nature Technology 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年07月07日管线快照
无数据报导
登录后保持更新
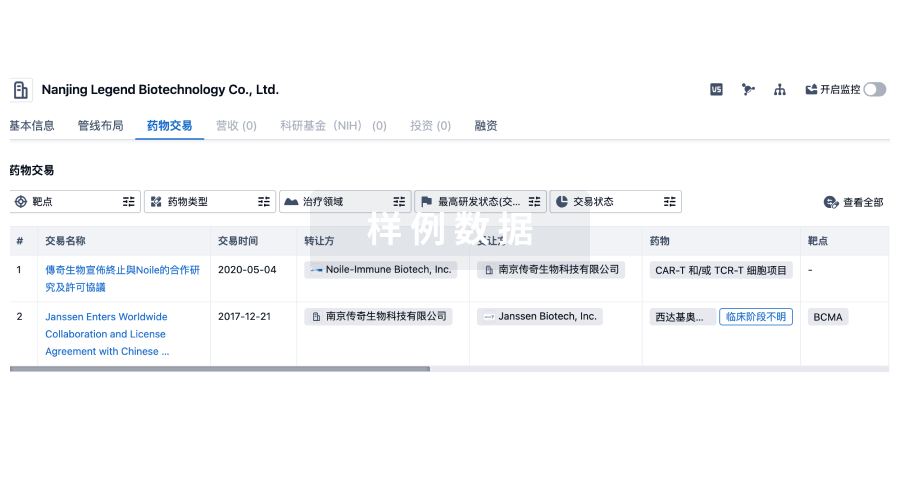
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
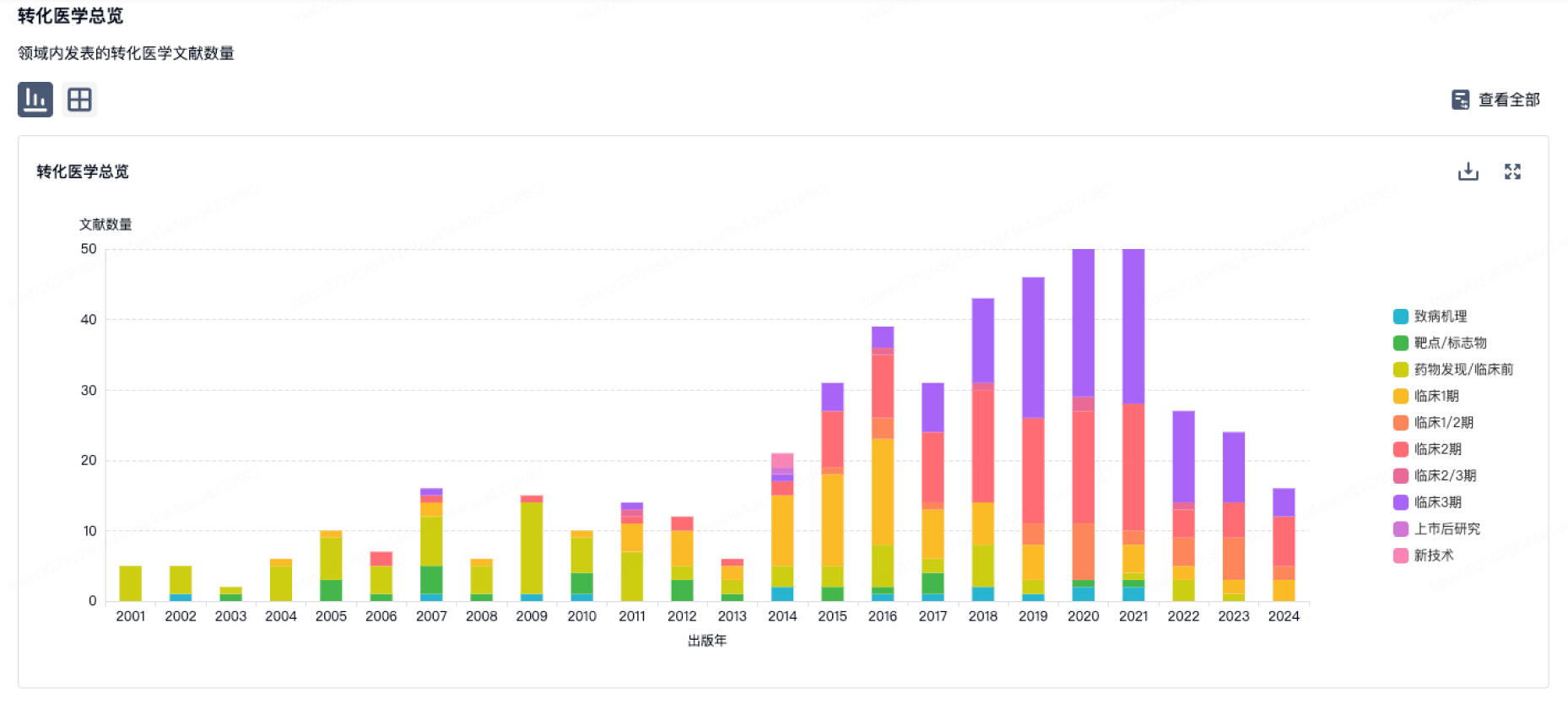
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
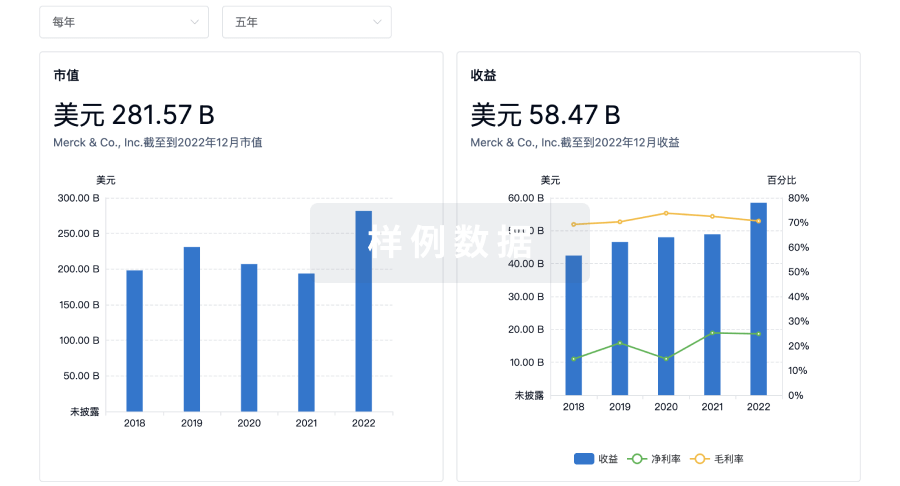
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
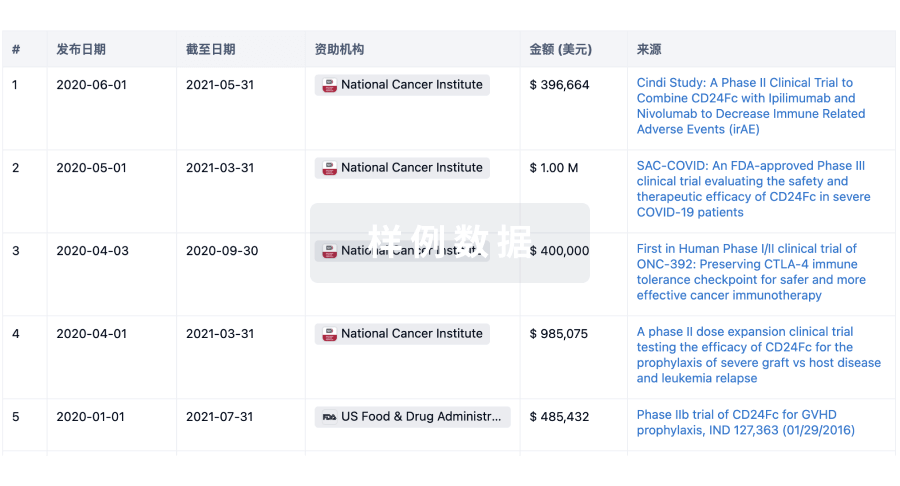
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用