预约演示
更新于:2025-05-07
PLXNA1
更新于:2025-05-07
基本信息
别名 NOV、plexin A1、Plexin-A1 + [3] |
简介 Coreceptor for SEMA3A, SEMA3C, SEMA3F and SEMA6D. Necessary for signaling by class 3 semaphorins and subsequent remodeling of the cytoskeleton. Plays a role in axon guidance, invasive growth and cell migration. Class 3 semaphorins bind to a complex composed of a neuropilin and a plexin. The plexin modulates the affinity of the complex for specific semaphorins, and its cytoplasmic domain is required for the activation of down-stream signaling events in the cytoplasm. Acts as coreceptor of TREM2 for SEMA6D in dendritic cells and is involved in the generation of immune responses and skeletal homeostasis. |
关联
100 项与 PLXNA1 相关的临床结果
登录后查看更多信息
100 项与 PLXNA1 相关的转化医学
登录后查看更多信息
0 项与 PLXNA1 相关的专利(医药)
登录后查看更多信息
192
项与 PLXNA1 相关的文献(医药)2025-02-01·YAKUGAKU ZASSHI
Elucidating the Pathophysiology of Various Diseases by Investigating the Role of Molecules in Brain Wiring
Review
作者: Yukawa, Kazunori
2025-01-01·Life Sciences
Semaphorins and its receptors: Emerging cellular biomarkers and therapeutic targets in autoimmune and inflammatory disorders
Review
作者: Qamar, Tooba ; Misra, Durga Prasanna ; Kar, Susanta
2024-12-01·Kidney Medicine
Circulating Protein and Metabolite Correlates of Histologically Confirmed Diabetic Kidney Disease
Article
作者: Surapaneni, Aditya ; Lopez-Silva, Carolina ; Waikar, Sushrut S ; Schmidt, Insa M ; Grams, Morgan E ; Rhee, Eugene P ; Srivastava, Anand ; Upadhyay, Dhairya ; Stillman, Isaac E ; Palsson, Ragnar
7
项与 PLXNA1 相关的新闻(医药)2025-04-23
·医药速览
细胞外蛋白(包括分泌蛋白与跨膜蛋白)在多细胞生物的生理调控中具有重要作用,其通过介导细胞间通讯协调复杂生理过程。病理状态下,细胞外蛋白的异常表达或修饰与癌症、神经退行性疾病及心血管疾病等发生发展密切相关。值得注意的是,超过60%的FDA批准药物靶向细胞外蛋白,因此系统解析细胞外蛋白质组对阐明生物过程分子机制、揭示疾病病理基础及开发靶向治疗策略具有重要意义。近年来,尽管邻近标记酶(proximity labeling enzymes)和小分子光催化剂技术的发展促进了细胞外蛋白质组的解析,但这些方法的体内应用仍存在局限性。辣根过氧化物酶(Horseradish Peroxidase, HRP)的标记依赖具有细胞毒性的过氧化氢(图1a),限制了其在活体动物中的直接使用;而生物素连接酶(如 BioID和 TurboID),虽然可以实现活体细胞内蛋白的标记,却因细胞外微环境中ATP浓度不足导致胞外标记效率受限。此外,内源性生物素或过氧化氢均可能出现较高的背景干扰。可见光在活体组织中的穿透深度有限,光催化剂在体内的应用也面临挑战。2025年3月15日,清华大学药学院秦为课题组在Nature Communications杂志上发表了题为 “Spatiotemporally resolved mapping of extracellular proteomes via in vivo-compatible TyroID” 的研究论文。该研究开发了一种无毒的邻近标记技术TyroID,实现了活体内细胞外蛋白质组的高时空分辨率解析,该技术成功应用于活体肿瘤异种移植模型HER2邻近蛋白动态解析、血浆蛋白代谢追踪及小鼠海马区特异性蛋白质组标记,解决了传统邻近标记技术在解析细胞外蛋白质组的活体兼容性问题。科研内容此研究发展了一种新型活体邻近标记技术TyroID(图1b),用于实现细胞外蛋白质组的时空动态解析。TyroID 技术的核心是基于植物或细菌来源的酪氨酸酶(tyrosinase),其可以将生物正交的苯酚底物氧化为高反应活性的邻醌(o-quinone),特异性标记邻近蛋白中半胱氨酸、赖氨酸及组氨酸等亲核氨基酸。与传统邻近标记技术相比,TyroID无需依赖过氧化氢或可见光等外源激活条件,仅需添加酚类底物即可完成原位标记。图1. (a)过氧化物酶(如 APEX 和 HRP)标记示意图;(b)TyroID标记示意图针对常规探针AP易渗入胞内的缺陷,团队开发了膜非通透型探针"AxxP"——通过引入四聚乙二醇连接臂阻断跨膜扩散。实验证明,75nM abmTYR联合50μM AxxP能在30分钟内实现乳腺癌细胞表面蛋白的特异性标记,与膜标志物GLUT1呈高度共定位,且维持生理状态下的蛋白空间分布。基于二甲基化定量蛋白质组学,TyroID系统成功鉴定315个高置信度细胞外蛋白。通过super-TOPP-ABPP技术解析的修饰位点均位于蛋白胞外域,证明该方法具备解析跨膜蛋白拓扑结构的独特能力。该研究进一步将TyroID应用于HER2受体邻近蛋白组解析,通过抗体靶向策略识别出LGALS1、FUS等HER2互作蛋白,并经Co-IP实验验证其结合强度与经典互作蛋白TFRC相当。最后,基于TyroID无毒、快速标记的优势,团队探索了其在活体中的应用,这是基于过氧化物酶和可见光激活的邻近标记方法难以实现的。为此,作者设计了三项独立的活体实验,以验证TyroID在活体小鼠中应用的有效性:1)活体肿瘤中HER2邻近蛋白的分析,鉴定出内质网ATP酶(VCP)、钙调理蛋白2(CNN2)及分选连接蛋白9(SNX9)等在细胞培养实验中未发现的HER2邻近蛋白;2)定量分析44种血浆蛋白的turnover速率,揭示Apoc3、Apoa4和Apoe等载脂蛋白的高代谢特性;3)海马区原位标记丛蛋白A1(Plxna1)、神经软骨蛋白(Ncdn)及神经元膜糖蛋白M6-a(Gpm6a)等脑特异性蛋白。图2. TyroID在活体内的应用综上,TyroID通过酶促生成邻醌实现无毒、快速的细胞外标记,突破了传统技术对H₂O₂或可见光的依赖,为活体蛋白质组动态解析提供了创新工具。其在肿瘤微环境研究、血浆蛋白周转监测及神经生物学领域的应用,展现了广阔的研究潜力。欢迎感兴趣的朋友进群讨论,群二维码如下:推文用于传递知识,如因版权等有疑问,请于本文刊发30日内联系医药速览。原创内容未经授权,禁止转载至其他平台。有问题可发邮件至yong_wang@pku.edu.cn获取更多信息。©2021 医药速览 保留所有权利往期链接“小小疫苗”养成记 | 医药公司管线盘点 人人学懂免疫学| 人人学懂免疫学(语音版)综述文章解读 | 文献略读 | 医学科普|医药前沿笔记PROTAC技术| 抗体药物| 抗体药物偶联-ADC核酸疫苗 | CAR技术| 化学生物学温馨提示医药速览公众号目前已经有近12个交流群(好学,有趣且奔波于医药圈人才聚集于此)。进群请扫描上方二维码,备注“姓名/昵称-企业/高校-具体研究领域/专业”,此群仅为科研交流群,非诚勿扰。简单操作即可星标⭐️医药速览,第一时间收到我们的推送①点击标题下方“医药速览” ②至右上角“...” ③点击“设为星标
2024-11-14
·生物谷
外泌体是细胞分泌的纳米级囊泡,它们在细胞间通讯中扮演着关键角色,并参与多种生理和病理过程。这些纳米级别的结构不仅能够携带蛋白质、脂质、以及核酸等生物分子,还能穿越生物屏障,如血脑屏障,为靶向药物递送和基因治疗提供了一个全新的平台。而工程化外泌体技术因能够通过基因或化学手段改善外泌体生物活性、稳定性以及靶向性,在外泌体药物递送及治疗应用中至关重要。其中通过基因工程方式进行内源性工程化外泌体改造是工程化外泌体应用的基石。
近日恩泽康泰工程化外泌体底层技术平台取得重大突破,其研究成果” PlexinA1 (PLXNA1) as a novel scaffold protein for the engineering of extracellular vesicles”登陆外泌体学术顶刊《Journal of Extracellular Vesicles》,代表着恩泽康泰成为全球少有的具有自己工程化外泌体操作系统的企业。下面一起来了解一下这项研究内容:
1. 研究背景
在过去的十年间,基于外泌体的创新疗法因为独有的优势而备受关注。这些优势包括有较低的免疫源性和良好的安全性(Gehrmann et al., 2014; Zhu et al., 2017) 、一定程度的组织选择性(Hoshino et al., 2015),以及能够实现跨血脑屏障的药物递送等(Chen et al., 2016; Yang et al., 2015)。然而随着研究的不断深入,研究人员逐渐认识到外泌体的成药之路并非一片坦途。
首先,外泌体虽然携带有大量来自母细胞的生物分子,但是当我们聚焦到某个特定生物分子的含量时,就会发现其平均拷贝数之低,到了令人发指的程度。例如,看家基因的mRNA在天然外泌体中,大约每10000个外泌体中才有1个拷贝。而我们所熟知的在外泌体中富集的四跨膜蛋白,在每一个外泌体中也不过4到5个的水平(见表1)。
表1、天然外泌体内含物的平均拷贝数
不仅如此,经静脉注射的外泌体在外周血中的循环时间太短,半衰期通常在30分钟左右(Imai et al., 2015; Parada et al., 2021)。而且生物学分布受限,无论是什么细胞来源的外泌体都显示出以肝脏为主的被动靶向(Morishita et al., 2015; Wiklander et al., 2015)。除此之外,量产工艺不成熟,缺乏有效的表征手段和质量标准都限制了外泌体疗法的发展速度。面对这样棘手的问题,大家是不是也想长叹一声“难办”。
为了克服上述的问题,科研人员进行了一系列的尝试。说到这里就不得不提到一个被寄予厚望技术路径——外泌体的工程化改造。工程化改造是指通过基因或者化学手段,改变外泌体原有的组成,从而使其特定的属性,如生物活性、稳定性以及靶向性等,向有利于应用的方向改善。
工程化改造外泌体
(图片来源:Front Bioeng Biotechnol. 2023 Jan 24;11:1100310.)
2. 科学问题
外泌体的工程化改造可以通过不同的方式来实现。这其中通过与支架蛋白(内源蛋白元件)融合表达的方式,将目的蛋白(protein of interest,POI)定向导入工具细胞所分泌的外泌体中的方法被称为生成前的蛋白质改造(pre-mature protein modification)或内源蛋白质改造(Endogenous protein modification)。因为这种改造是对细胞而非外泌体进行的,因此可以通过构建单克隆细胞株的方式实现稳定大批量的重复制备。此外,根据需要还可以将POI展示在外泌体的胞外或者包裹在囊泡内部。因此这种改造方式是目前使用最为广泛,而且可实现的功能也最为丰富的一类。
目前,常见的外泌体支架蛋白分为三类,分别是四跨膜蛋白(CD63,CD9,CD81)、膜周蛋白(MFGE8),以及I型跨膜蛋白(Lamp2b,PTGFRN)三类(Choi et al., 2022)。然而,现有的这些支架蛋白或多或少都存在着一些不足之处,并不能彻底满足外泌体工程化改造的诸多需求。
例如,四跨膜蛋白的N端和C端均位于胞内,当需要在外泌体的胞外展示POI的时候只能从胞外的loop结构进行融合改造,加大了分子设计和验证的难度;膜周蛋白MFGE8虽然在外泌体上表达量很高,但是它和膜之间的作用力并不强,一旦融合分子量较大的POI后,在外泌体上的含量水平就会迅速降低;Lamp2b作为I型跨膜支架蛋白的主要代表,只适合在N端(即胞外段)融合POI,如果在C端进行融合就会破坏其YXXΦ信号的功能,从而影响融合蛋白向外泌体中分拣的能力。
外泌体工程化改造的实现方式,
图片来源(Choi et al., 2022)
为了让外泌体工程化改造技术能够更好得服务于外泌体创新疗法的开发,筛选新型支架蛋白的努力从未停止。而此前筛选外泌体支架蛋白的标准基本包括以下四个:
1.支架蛋白本身在外泌体上的表达水平越高越好;
2.支架蛋白本身的分子量不能够太大;
3.支架蛋白上需要有适合进行改造的位点;
4.支架蛋白本身不应该具有明显的生物学活性。
然而,能够同时满足上述四个条件的候选分子数极其有限。Zheng及其团队在2023年发表的研究中,将支架蛋白的分子量上限设为130kDa,最终找到的新支架蛋白仍然以四跨膜蛋白超家族为主(Zheng et al., 2023)。因此,在如此苛刻的筛选标准下,想要找到性能更好的支架蛋白,可能性十分渺茫。那么有没有可能通过放宽筛选标准的方法,找到更多更适合用于外泌体工程化改造的支架蛋白呢?为了回答这个问题,我们开展了本项研究。
3. 研究内容
在开展研究前,我们改变了此前的研究策略,将支架蛋白的筛选标准进行了大幅度的精简,从之前的四个减少到以下两个:
1.必须是具有外泌体主动分拣能力的I型跨膜蛋白;
2.在该蛋白的N端以及C端进行截短或融合等改造不会显著影响该蛋白向外泌体主动分拣的能力。
这一标准对于支架蛋白的分子量、在外泌体中的表达水平,以及是否具有生物学活性等不作要求。我们预计基于这两条标准,能够将大量原本不被考虑的蛋白纳入到候选分子列表中。我们之所以在第一个条件中,排除了I型跨膜蛋白之外的其他候选分子,一方面是为了避免候选分子列表过于庞大,另一方面是希望能够找到既能够用于胞外展示也能够用于胞内装载的理想的支架蛋白。
3.1、外泌体蛋白质质谱
首先,我们基于蛋白质质谱对不同蛋白的外泌体分拣能力进行了初步评估。质谱结果显示,丛蛋白A1(PLXNA1)与经典的外泌体富集蛋白(MFGE8、CD81、PTGFRN,以及SDCBP等)类似,其相对丰度在细胞裂解液、初纯外泌体(UC),和高纯外泌体(DGC)之中依次呈现出由低到高的趋势,而与外泌体无关的杂蛋白(CANX,histone)呈现的趋势则刚刚相反。并且该蛋白的在外泌体中相比细胞裂解液中的含量提高的倍数要显著高于几乎所有的经典外泌体富集蛋白,仅次于MFGE8。这一现象也得到了Westernblot的证实。这些结果提示PLXNA1满足我们所提出的第一条标准。
3.2、PLXNA1的截短和融合改造并不影响其外泌体分拣能力
按照之前的筛选标准,PLXNA1作为外泌体支架蛋白存在天然的劣势。首先,其N端和C端各有一个具有生物学功能的结构域。其次,该蛋白的分子量很大约为211kDa。所以全长的PLXNA1并不适合作为外泌体支架蛋白。于是我们开发了一种基于Westernblot和纳米流式的方法,并对PLXNA1的截短体进行了评价结果显示该蛋白的多个截短体都具有较高的外泌体分拣能力,这表明PLXNA1也同样满足我们提出的第二条筛选标准。
3.3、PLXNA1截短体是优秀的外泌体支架蛋白
确定了PLXNA1截短体能够作为外泌体支架蛋白使用后,我们将其与已知的I型跨膜支架蛋白(Lamp2b和PTGFRN)进行了头对头的比较。结果显示PLXNA1相比这两个蛋白表现出了明显的载量优势。
不仅如此,PLXNA1因其在胞外端和胞内端都能够融合表达POI,且不影响外泌体分拣能力,因此可以胜任多种工程化改造的用途。例如,可以用于内源mRNA的装载,以及胞外和/或胞内的蛋白展示等。而且,无论展示在胞外还是胞内,都不影响POI自身的生物学活性。这对于工程化外泌体的设计而言提供了极大的便利。
3.4、基于ALFAtag/NbALFA的通用外泌体
在本研究的最后,我们基于ALFAtag/NbALFA系统构建了一个通用的工程化外泌体。在这种通用的工程化外泌体的表面,我们将NbALFA纳米抗体融合表达在PLXNA1的N端。经过体外孵育,带有ALFAtag标签的重组蛋白能够与之结合从而固定在外泌体的表面。这种标记方法不仅具有很高的标记效率,而是能够在多个条件下长时间稳定保存。甚至可以在外泌体表面标记靶向特定抗原的抗体片段,从而实现对抗原表达阳性细胞的靶向递送。
4. 研究意义
本研究对于外泌体工程化改造技术及其应用有着十分重要的意义,其亮点可以简单总结如下:
1. 提出了一种全新的支架蛋白筛选标准,极大地拓展了候选支架蛋白的选择范围,为后续的研究提供了新的路径;
2. 鉴定了新型的外泌体支架蛋白PLXNA1,该蛋白不仅具有极强的外泌体分拣能力,而且还允许在外泌体的胞外和腔内分别或同时融合表达POI,为外泌体的工程化改造提供了强有力新的选择;
3. 发现了一个有助于蛋白分拣到外泌体中的关键基序,对于工程化外泌体的分子设计提供新的参考;
4. 开发了一种基于ALFAtag/NbALFA的通用外泌体,有望成为工程化外泌体载体或药物开发的重要工具。
点评专家:尹航教授
专家简介:清华大学校学术委员会委员、校科技伦理委员会委员,药学院原副院长,兼任Bioorganic & Medicinal Chemistry Letters (Elsevier)主编、Journal of Extracellular Vesicle (Wiley)责任主编、Cell Chemical Biology (Cell Press)编委、中国细胞外囊泡研究与应用学会顾问、中国生物医药产业链创新与转化联盟常务理事及重大需求专委会主任委员、全国中药标准化技术委员会委员等。尹航教授团队在本领域一流期刊发表研究论文超过150篇;科研成果已申请专利超20项,并有多项成果转化。
尹航教授先后入选国家和中国科协海智计划特聘专家、获得国家自然基金委杰出青年科学基金、北京市卓越青年科学家奖、吴阶平-保罗·杨森医学药学奖、中国药学会科学技术一等奖、教育部自然科学二等奖、美国化学学会大卫·罗伯特森杰出药物化学家奖、美国国家科学基金会杰出青年教授奖、美国癌症研究学会格特魯德·B·埃利恩奖、中美化学及化学生物学教授联合会OKeanos-CAPA资深科学家奖、悉德尼·金梅尔学者奖等奖项。
点评内容:
近几年,无论是国内外的学界还是产业界,对细胞外囊泡(EVs)的关注度一直处于持续上升的态势。关于EVs创新疗法的研究,更是多次刊登在高水平的学术期刊上。这些研究给我们展示了EVs作为一种药物或者药物递送系统,相比现有疗法(modality)的种种优势以及丰富的可能性。但与此同时,我们也在这个过程中认识到了EVs疗法距离取得重大的临床突破,还有一系列的技术瓶颈有待克服。
此次,北京恩泽康泰团队针对EVs工程化改造中的一个关键问题,即支架蛋白筛选和改造的底层逻辑展开了研究,并取得了重要进展。首先,这项研究回答了为何在相当长的一段时间内,可用于EVs工程化改造的新支架蛋白的发掘陷入了低谷。另一方面,他们通过系统性的研究,证明简化原有的筛选标准有助于提高筛选到优质支架蛋白的成功率。在该研究的最后,他们基于新鉴定的支架蛋白(PLXNA1)开展了大量且深入的实验,向我们充分展示了工程化外泌体作为药物载体的应用潜力。
这项研究可以说代表了外泌体工程化技术的发展趋势,对于未来该领域的相关研究有着较大的指导意义。对于中国本土的企业能够凭借自己的努力取得这样的成绩,达到与国际接轨的水平,我也感到非常的高兴。希望我们国内学界和产业界的同行能够携手努力,让中国站在国际细胞外囊泡研究、应用,以及转化的最前沿。
参考文献:
[1]Hang Zhao, Zhi Li., et al. (2024). PlexinA1 (PLXNA1) as a novel scaffold protein for the engineering of extracellular vesicles. J Extracell Vesicles, 13:e70012.
[2]Chen, C.C., Liu, L., Ma, F., Wong, C.W., Guo, X.E., Chacko, J.V., Farhoodi, H.P., Zhang, S.X., Zimak, J., Segaliny, A., et al. (2016). Elucidation of Exosome Migration across the Blood-Brain Barrier Model In Vitro. Cell Mol Bioeng 9, 509-529.
[3]Chevillet, J.R., Kang, Q., Ruf, I.K., Briggs, H.A., Vojtech, L.N., Hughes, S.M., Cheng, H.H., Arroyo, J.D., Meredith, E.K., Gallichotte, E.N., et al. (2014). Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc Natl Acad Sci U S A 111, 14888-14893.
[4]Choi, H., Yim, H., Park, C., Ahn, S.H., Ahn, Y., Lee, A., Yang, H., and Choi, C. (2022). Targeted Delivery of Exosomes Armed with Anti-Cancer Therapeutics. Membranes (Basel) 12.
[5]Gehrmann, U., Naslund, T.I., Hiltbrunner, S., Larssen, P., and Gabrielsson, S. (2014). Harnessing the exosome-induced immune response for cancer immunotherapy. Semin Cancer Biol 28, 58-67.
[6]Hoshino, A., Costa-Silva, B., Shen, T.L., Rodrigues, G., Hashimoto, A., Tesic Mark, M., Molina, H., Kohsaka, S., Di Giannatale, A., Ceder, S., et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329-335.
[7]Imai, T., Takahashi, Y., Nishikawa, M., Kato, K., Morishita, M., Yamashita, T., Matsumoto, A., Charoenviriyakul, C., and Takakura, Y. (2015). Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J Extracell Vesicles 4, 26238.
[8]Morishita, M., Takahashi, Y., Nishikawa, M., Sano, K., Kato, K., Yamashita, T., Imai, T., Saji, H., and Takakura, Y. (2015). Quantitative analysis of tissue distribution of the B16BL6-derived exosomes using a streptavidin-lactadherin fusion protein and iodine-125-labeled biotin derivative after intravenous injection in mice. J Pharm Sci 104, 705-713.
[9]Parada, N., Romero-Trujillo, A., Georges, N., and Alcayaga-Miranda, F. (2021). Camouflage strategies for therapeutic exosomes evasion from phagocytosis. J Adv Res31, 61-74.
[10]Tian, Y., Ma, L., Gong, M., Su, G., Zhu, S., Zhang, W., Wang, S., Li, Z., Chen, C., Li, L., et al.(2018). Protein Profiling and Sizing of Extracellular Vesicles from Colorectal Cancer Patients via Flow Cytometry. ACS Nano 12, 671-680.
[11]Wei, Z., Batagov, A.O., Schinelli, S., Wang, J., Wang, Y., El Fatimy, R., Rabinovsky, R., Balaj, L., Chen, C.C., Hochberg, F., et al.(2017). Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat Commun 8, 1145.
[12]Wiklander, O.P., Nordin, J.Z., O'Loughlin, A., Gustafsson, Y., Corso, G., Mager, I., Vader, P., Lee, Y., Sork, H., Seow, Y., et al. (2015). Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles4, 26316.
[13]Yang, T., Martin, P., Fogarty, B., Brown, A., Schurman, K., Phipps, R., Yin, V.P., Lockman, P., and Bai, S. (2015). Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res 32, 2003-2014.
[14]Zheng, W., Radler, J., Sork, H., Niu, Z., Roudi, S., Bost, J.P., Gorgens, A., Zhao, Y., Mamand, D.R., Liang, X., et al. (2023). Identification of scaffold proteins for improved endogenous engineering of extracellular vesicles. Nat Commun 14, 4734.
[15]Zhu, X., Badawi, M., Pomeroy, S., Sutaria, D.S., Xie, Z., Baek, A., Jiang, J., Elgamal, O.A., Mo, X., Perle, K., et al. (2017). Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J Extracell Vesicles 6, 1324730.
本文仅用于学术分享,转载请注明出处。若有侵权,请联系微信:bioonSir 删除或修改!
信使RNA基因疗法
2024-10-21
MONTREAL, Canada I October 21, 2024 I
Find Therapeutics Inc., a clinical-stage biopharmaceutical company focused on the development of innovative therapies for autoimmune diseases, today announced initiation of dosing in its Phase 1 clinical study of FTX-101 in healthy volunteers. FTX-101 is a first-in-class therapeutic peptide that modulates the Plexin A1 / Neuropilin 1 transmembrane receptor complex in the brain to promote remyelination. Following completion of the Phase 1 clincal study, Find plans to investigate the use of FTX-101 in individuals with Chronic Optic Neuropathy – a long-term condition due to demyelination in the visual tracts of the brain and optic nerve, with serious effects on visual acuity and perception.
The Phase 1 first-in-human study will assess the safety, tolerability, and pharmacokinetics of FTX-101 when administered in healthy volunteers. The study is being initiated after the US Food and Drug Administration authorized initiation of the study earlier this year. The clinical study consists of two parts. Part 1 is a randomized, double-blind, placebo-controlled, single ascending dose (SAD) study comprised of 40 subjects receiving FTX-101 or placebo while Part 2 is a randomized, double-blind, placebo-controlled, multiple ascending dose (MAD) study that will enroll 24 subjects receiving FTX-101 or placebo.
“The initiation of this study is a significant milestone for both our company and individuals who suffer with Chronic Optic Neuropathy,” said Philippe Douville, CEO of Find. “In this study, we will build on the large body of preclinical evidence demonstrating FTX-101’s effects in models of autoimmune disease. A therapy of this type would represent a major step forward in finding the first treatment to improve the lives of people afflicted with Chronic Optic Neuropathy. We look forward to sharing data from this study in the first half of 2025.”
About FTX-101
Find’s lead compound, FTX-101, is a first-in-class remyelinating agent that aims to restore vision to people suffering from Chronic Optic Neuropathy. FTX-101 is a rationally designed therapeutic peptide that targets Plexin A1 and Neuropilin 1, a receptor complex present in the brain that has been shown to be involved in the migration and differentiation of oligodendrocyte precursor cells into myelinating oligodendrocytes. Compelling preclinical data for FTX-101 in demyelinating models show strong myelin repair activity.
About Find
Find Therapeutics is clinical-stage biopharmaceutical company dedicated to the development of next generation therapies to treat inflammatory autoimmune diseases. The company was launched in 2020 and is supported with investments from CTI Life Sciences, adMare BioInnovations, Domain Therapeutics and Investissment Québec and an exclusive license from SATT-Conectus and the University of Strasbourg on a technology and related know-how. Visit
www.findtherapeutics.com
for more details about Find Therapeutics.
SOURCE:
Find Therapeutics
临床1期引进/卖出
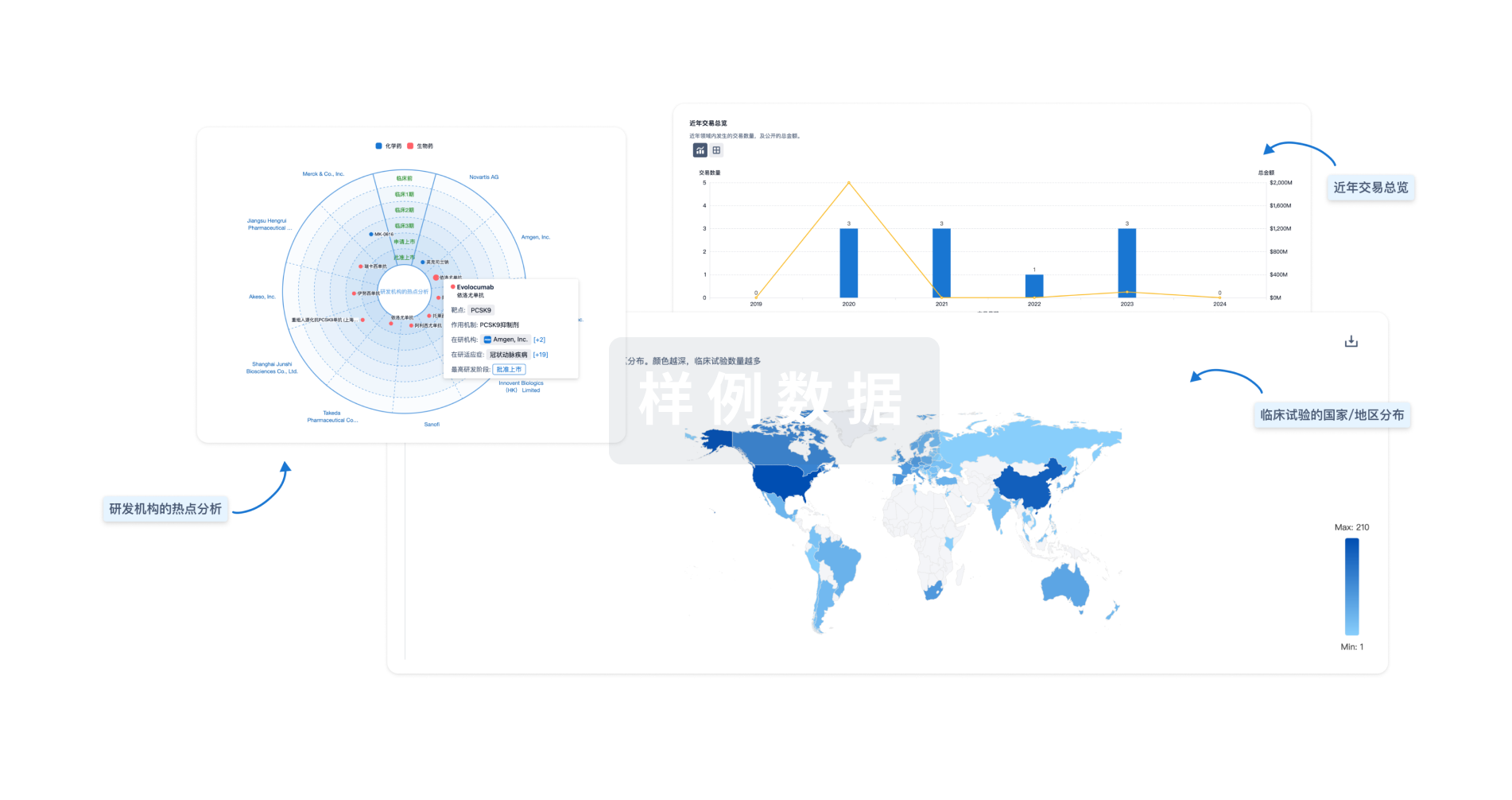
分析
对领域进行一次全面的分析。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用