预约演示
更新于:2026-03-11
HER2 inhibitors(Boehringer Ingelheim)
更新于:2026-03-11
概要
基本信息
药物类型 小分子化药 |
别名 BI-1622、BI-4142 |
作用方式 抑制剂 |
作用机制 HER2 exon 20抑制剂(ERBB2-受体蛋白酪氨酸激酶20外显子突变抑制剂) |
在研适应症 |
非在研适应症- |
非在研机构- |
权益机构- |
最高研发阶段临床前 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |
结构/序列
分子式C28H27N9O2 |
InChIKeyJKBYBPNRTMHEGS-UHFFFAOYSA-N |
CAS号2682003-36-5 |
关联
100 项与 HER2 inhibitors(Boehringer Ingelheim) 相关的临床结果
登录后查看更多信息
100 项与 HER2 inhibitors(Boehringer Ingelheim) 相关的转化医学
登录后查看更多信息
100 项与 HER2 inhibitors(Boehringer Ingelheim) 相关的专利(医药)
登录后查看更多信息
180
项与 HER2 inhibitors(Boehringer Ingelheim) 相关的文献(医药)2026-01-01·CELL BIOCHEMISTRY AND FUNCTION
Targeting Estrogen Pathways in Breast Cancer: A Review of Current Therapies and Emerging Strategies
Review
作者: Bonde, Chandrakant ; Dabhi, Maitri ; Parmar, Ishvarchandra
ABSTRACT:
Breast cancer continues to be the most commonly diagnosed cancer among women worldwide, with estrogen receptor‐positive (ER +) types making up the largest portion. At the heart of this subtype is the estrogen signaling pathway, especially the estrogen receptor alpha (ERα), which plays a major role in the development, growth, and response to these cancers. This review takes a close look at the structure and function of estrogen receptors and how they influence cancer progression. We explore current treatment strategies—including well‐known drugs such as Tamoxifen and Fulvestrant, as well as aromatase inhibitors—and explain how these therapies work and why resistance sometimes develops. This review also dives into newer, more targeted options, such as HER2 inhibitors, CDK4/6 blockers, and drugs that interfere with the PI3K/AKT/mTOR pathway. These treatments are changing the game for many patients. In addition, the review highlights exciting progress in drug design, showing how researchers are improving the precision and effectiveness of cancer medications through innovations in medicinal chemistry. Antibody‐drug conjugates (ADCs) are being developed to deliver powerful drugs directly to cancer cells with fewer side effects. This article also looks at emerging approaches, like oral estrogen receptor degraders (SERDs), combination therapies, and precision medicine techniques that tailor treatment based on each patient's unique genetic profile. Altogether, these developments represent a major step forward in our understanding and treatment of ER+ breast cancer.
2026-01-01·Asian Pacific journal of cancer prevention : APJCP
Comprehensive Molecular Docking and Molecular Dynamics Reveal Inhibitors of HER2 L755S, T798I, and T798M based on a Large Database of Curcumin Derivatives
Article
作者: Gusrin, Mantiqa Syafa Duvadillan ; Utomo, Rohmad Yudi ; Larasati, Yonika Arum
OBJECTIVE:
This study presents a methodology employing virtual screening to identify curcumin derivatives with selective affinity for the HER2 mutations L755S, T798I, and T798M.
METHODS:
Curcumin derivatives were retrieved from the ChEMBL database and filtered using KNIME. HER2 mutations were modeled in silico using MOE software with PDB ID 3RCD. Molecular docking and dynamics simulations were conducted to screen high-affinity compounds and evaluate binding interactions.
RESULT:
From 505 curcumin derivatives, the RDKit module implemented in KNIME successfully filtered 317 compounds. Subsequent molecular docking against wild-type HER2 identified 100 curcumin derivatives with low docking scores, among which the top 20 compounds exhibited better binding affinities than Lapatinib. Further molecular docking screening against the three HER2 mutations identified five lead compounds with the lowest docking scores. Molecular docking and molecular dynamics simulation revealed critical binding interactions with residues essential for kinase domain stability. Chemical structural analysis revealed key modifications, such as geranyl and tripeptide modifications. CHEMBL3758656 and CHEMBL3827366, two curcumin derivatives, demonstrated consistent binding across HER2 mutations and a favorable ADMET profile.
CONCLUSION:
This study successfully identified CHEMBL3758656 and CHEMBL3827366 as promising HER2 inhibitors through comprehensive virtual screening. Their high binding affinity against L755S, T798I, and T798M mutations and favorable ADME and toxicity properties underscore their potential as alternative therapeutics for HER2-positive breast cancer.
2026-01-01·ChemistryOpen
Design, Synthesis, Bioevaluation, and Bioinformatics Study of 5‐Benzylidene Hydantoin Derivatives as Novel Tyrosine Kinase Inhibitors
Article
作者: Hidayat, Ika Wiani ; Al‐Anshori, Jamaludin ; Naufal, Muhammad ; Hermawati, Elvira ; Danova, Ade
Tyrosine kinases regulate cellular growth, differentiation, and metabolism, and their dysregulation is implicated in malignancies, making them therapeutic targets. This study synthesizes novel 5‐benzylidene hydantoin derivatives (24–38) via benzylation and condensation, characterized by nuclear magnetic resonance (NMR), mass spectrometry, and fourier‐transform infrared (FTIR). Anticancer activity was evaluated against eight receptor tyrosine kinases at 10 μM. Six compounds—24 (34%), 25 (45%), 28 (57%), 32 (60%), 34 (49%), and 38 (56%)—show moderate HER2 inhibition (%enzyme activity ≤ 60%). Compound 38 additionally inhibits VEGFR2 (27%), PDGFRα (32%), and PDGFRβ (25%). Molecular docking reveals interactions with HER2 residues Met801, Leu726, Leu852, Phe1004, Val734, and Leu796, suggesting a structural basis for selectivity. The HER2‐targeting derivatives demonstrate potential for development as novel HER2 inhibitors. Compound 38's multikinase inhibition resembles sunitinib, a clinically approved drug for renal cell carcinoma and gastrointestinal stromal tumors, highlighting its promise for broader kinase‐targeted therapy. These findings underscore the therapeutic relevance of the 5‐benzylidene hydantoin scaffold, warranting further optimization to enhance potency and selectivity against HER2 and other oncogenic kinases.
10
项与 HER2 inhibitors(Boehringer Ingelheim) 相关的新闻(医药)2025-10-17
An alliance of scientists at the Broad Institute and Bayer Pharmaceuticals have developed a drug candidate, sevabertinib, that could be a new treatment for a group of lung cancer patients who have few options today.
In a new study published in
Cancer Discovery
, the team described their efforts to develop sevabertinib. They tested the compound in various lung cancer models and showed its potential to treat non-small cell lung cancers that harbor certain mutations in the
ERBB2
gene, which encodes the HER2 protein. These mutations occur in 2 to 4 percent of patients with non-small cell lung cancer, or roughly 40,000 to 50,000 people diagnosed globally each year. These patients tend to be women, including those who are younger, have never smoked, and have a poor prognosis.
The study also reported data from two participants in Bayer’s phase 1/2 clinical trial of the compound. Based on these findings and other data from this ongoing clinical trial, the drug candidate is currently under
Priority Review at the FDA
, an expedited review of therapies that treat serious conditions. If approved, it would be the first FDA-approved cancer drug based on Broad discoveries, and the first new medicine from the Broad-Bayer oncology research alliance.
“This is an extraordinary time for the development of new treatments for patients with cancer and so many other diseases,” said study co-leader
Matthew Meyerson
, an institute member at the Broad, the Charles A. Dana Chair in Human Cancer Genetics at Dana-Farber Cancer Institute, and professor of genetics and medicine at Dana-Farber and Harvard Medical School. “Our work on sevabertinib is a real proof-of-concept that we can use genetic insights from cancer patients to develop next-generation cancer therapeutics. I’m hopeful that future collaborations between the Broad and industry partners like Bayer will continue to yield discoveries that could one day improve patient health.”
The work was also led by co-senior author
Heidi Greulich
, an institute scientist and senior group leader in the Cancer Program at the Broad, along with first author Franziska Siegel, research portfolio lead for oncology research at Bayer Pharmaceuticals.
In 2013, the Broad and Bayer launched a research alliance to find cancer drug targets and novel therapeutic approaches. One of the alliance’s first efforts was to search for potential drugs that target a specific mutation in lung cancer patients that Greulich, Meyerson, and Broad colleagues had studied nearly a decade earlier.
In 2005, the Broad team
reported
that some patients who didn’t respond to lung cancer drugs that targeted a protein called EGFR had a mutation in their tumors known as an “exon 20 insertion” — an extra bit of DNA tucked in at a specific location in the
EGFR
gene. They suggested that new drugs targeting this mutation could potentially benefit these patients.
In the new paper, the team showed that sevabertinib could target exon 20 insertions in both EGFR and in a related protein, HER2. The drug candidate inhibits an enzyme in the cell, called a “tyrosine kinase”, that helps promote cancer cell growth and survival, so the scientists theorized that blocking the enzyme could slow growth in tumors or shrink them.
Using cell models developed at Broad to carry the HER2 exon 20 insertions, the researchers demonstrated that the compound slowed the growth of cells by targeting the mutated HER2 protein, while largely not interfering with normal EGFR. Other results from experiments in cells suggest that the drug candidate might work in cases where other HER2 inhibitors might not, and may have activity in other cancers, such as breast and gastroesophageal that are marked by high HER2 levels.
In animal models carrying tumors with the HER2 mutations, the drug candidate shrank the tumors. And in two clinical trial participants who had already tried standard-of-care treatments, sevabertinib shrank lung tumors with the HER2 exon 20 insertion mutation.
“The development of this new drug candidate speaks to the power of collaboration, where we each bring different knowledge and skills,” said Greulich. “We faced challenges in this project, but through persistence and long-term combined efforts, we were able to find a potential new solution for a group of patients who need more options.”
Legal Disclaimer:
EIN Presswire provides this news content "as is" without warranty of any kind. We do not accept any responsibility or liability
for the accuracy, content, images, videos, licenses, completeness, legality, or reliability of the information contained in this
article. If you have any complaints or copyright issues related to this article, kindly contact the author above.
优先审批临床研究
2025-06-12
SOUTH SAN FRANCISCO, Calif.--(BUSINESS WIRE)--TheRas, Inc. d/b/a BBOT (the “Company”), a clinical-stage biopharmaceutical company focused on RAS-pathway malignancies, today announced the publication of preclinical data supporting the potential for BBO-10203 to provide therapeutic benefit across multiple tumor types. The publication, titled “BBO-10203 inhibits tumor growth without inducing hyperglycemia by blocking RAS-PI3Kα interaction” was published in the peer-reviewed journal Science.
These data describe the discovery and preclinical evaluation of BBO-10203, a first-in-class, orally available inhibitor that selectively blocks the interaction between RAS proteins and PI3Kα without impairing insulin signaling. By covalently binding to a unique cysteine in the RAS-binding domain of PI3Kα, BBO-10203 effectively disrupts oncogenic RAS-driven activation of the PI3Kα pathway across a range of tumor types, including those with mutations in KRAS, PIK3CA, and HER2 amplification, without inducing hyperglycemia. The compound showed broad antitumor activity in vitro and in vivo and demonstrated enhanced efficacy when combined with targeted therapies such as CDK4/6 inhibitors, ER antagonists, HER2 inhibitors, and KRASG12C inhibitors. These findings support the potential of BBO-10203 as a well-tolerated, mechanistically distinct therapeutic for PI3Kα- and RAS-driven cancers.
“Because the contribution of the second most mutated signaling pathway in human cancers remains underappreciated, we searched for an entirely novel molecular mechanism that is not encumbered by known metabolic liabilities to inhibit PI3Kα signaling,” said Pedro Beltran, PhD, Chief Scientific Officer of BBOT. “By blocking the crosstalk between RAS and PI3Kα in tumors, without interfering with physiological insulin signaling, we believe BBO-10203 represents a fundamentally differentiated approach with both biological and therapeutic promise.”
“This work stemmed from our goal to elucidate the structural basis of RAS:PI3Kα binding and therapeutically target this interaction,” said Dhirendra Simanshu, PhD, lead author and Principal Scientist at Frederick National Laboratory for Cancer Research (FNLCR). “Our findings show that targeting RAS-effector interactions is both structurally tractable and has potential across a range of cancers. BBO-10203 exemplifies a paradigm shift – rather than inhibiting RAS directly, it intercepts oncogenic signaling through effectors like PI3Kα, enabling tumor suppression while preserving essential physiological functions.”
BBO-10203 is currently being evaluated in our Phase 1 BREAKER-101 study (NCT06625775) in patients with locally advanced or metastatic HER2+ breast cancer, HR+/HER2- breast cancer, KRAS mutant advanced CRC, and KRAS mutant advanced NSCLC. The discovery of BBO-10203 was the result of a collaboration between the RAS Initiative at Frederick National Laboratory, Lawrence Livermore National Laboratory, and BBOT.
“This work is an excellent example of chemistry bringing clarity to biology,” said Frank McCormick, PhD, FRS, Chairman of the BBOT Board, Advisor to the National Cancer Institute’s RAS Initiative at Frederick National Laboratory for Cancer Research, and Professor of Tumor Biology and Cancer Research at UCSF. “The role of the RAS:PI3Kα interaction in cancer biology has long been suspected but challenging to pin down precisely. Now we understand which cancers depend on this interaction, some quite unexpected. With BBO-10203 now in the clinic, there's real hope that these discoveries will translate into meaningful benefit for many cancer patients.”
About BBOT
BBOT is a clinical-stage biopharmaceutical company advancing a next-generation pipeline of novel small molecule therapeutics targeting RAS and PI3Kα malignancies. Initially formed as a subsidiary of BridgeBio Pharma, Inc. (Nasdaq: BBIO), BBOT has the goal of improving outcomes for patients with cancers driven by the two most prevalent oncogenes in human tumors. For more information, visit bbotx.com.
临床1期
2025-04-28
CHICAGO —
Boehringer Ingelheim’s targeted lung cancer drug led to durable responses, according to the latest data from the early-stage trial.
The German drugmaker is studying zongertinib in lung cancer with HER2 mutations, which account for 2% to 4% of non-small cell lung cancer cases. As of Nov. 29, a 120 mg dose of the once-daily drug kept cancer at bay for a median of one year in 75 previously treated lung cancer patients. In addition, the median duration of response for those patients was 14 months.
The data from the Phase 1b Beamion LUNG-1 study were shared Monday at the American Association for Cancer Research’s annual meeting in Chicago and
published
the same day in
The
New England Journal of Medicine
.
Researchers also shared that 71% of patients in that cohort responded to treatment. The drug’s response rates and the duration of response data “looked really good,” Penn Medicine oncologist Charu Aggarwal told
Endpoints News
. Aggarwal was not involved in the study.
A key feature of zongertinib is that it targets HER2 while sparing EGFR, and it can be a potentially safer option for patients compared to other HER2 inhibitors. Unlike AstraZeneca and Daiichi Sankyo’s Enhertu, “it doesn’t have the risk of ILD [interstitial lung disease], which is a major risk for lung cancer patients,” John Heymach, chair of thoracic/head and neck medical oncology at MD Anderson Cancer Center, said in an interview in advance of the study’s presentation.
The most commonly reported adverse event in the study was mild diarrhea, though 17% of patients experienced grade 3 or higher drug-related adverse events like increased liver enzyme levels.
Boehringer
reported
an objective response rate of 71% in February when it announced that the FDA is expected to decide in the third quarter whether to approve zongertinib in advanced HER2-mutant non-small cell lung cancer based on the results of this study. The company is also running a Phase 3 trial called Beamion LUNG-2, studying zongertinib as a first-line option for patients with advanced HER2-mutated lung cancer.
When asked about the possibility of FDA delays, Boehringer oncology head Itziar Canamasas said the company is on track with the FDA’s decision date. “We’re monitoring the situation very closely,” she said.
临床结果临床1期临床3期AACR会议临床2期
100 项与 HER2 inhibitors(Boehringer Ingelheim) 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 非小细胞肺癌 | 临床前 | 奥地利 | - |
登录后查看更多信息
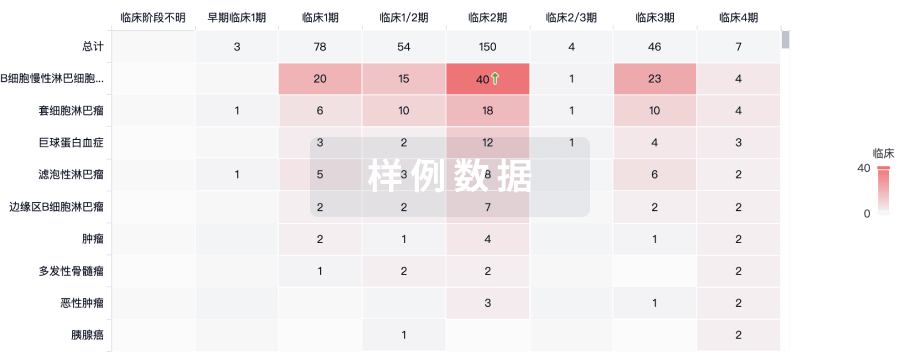
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
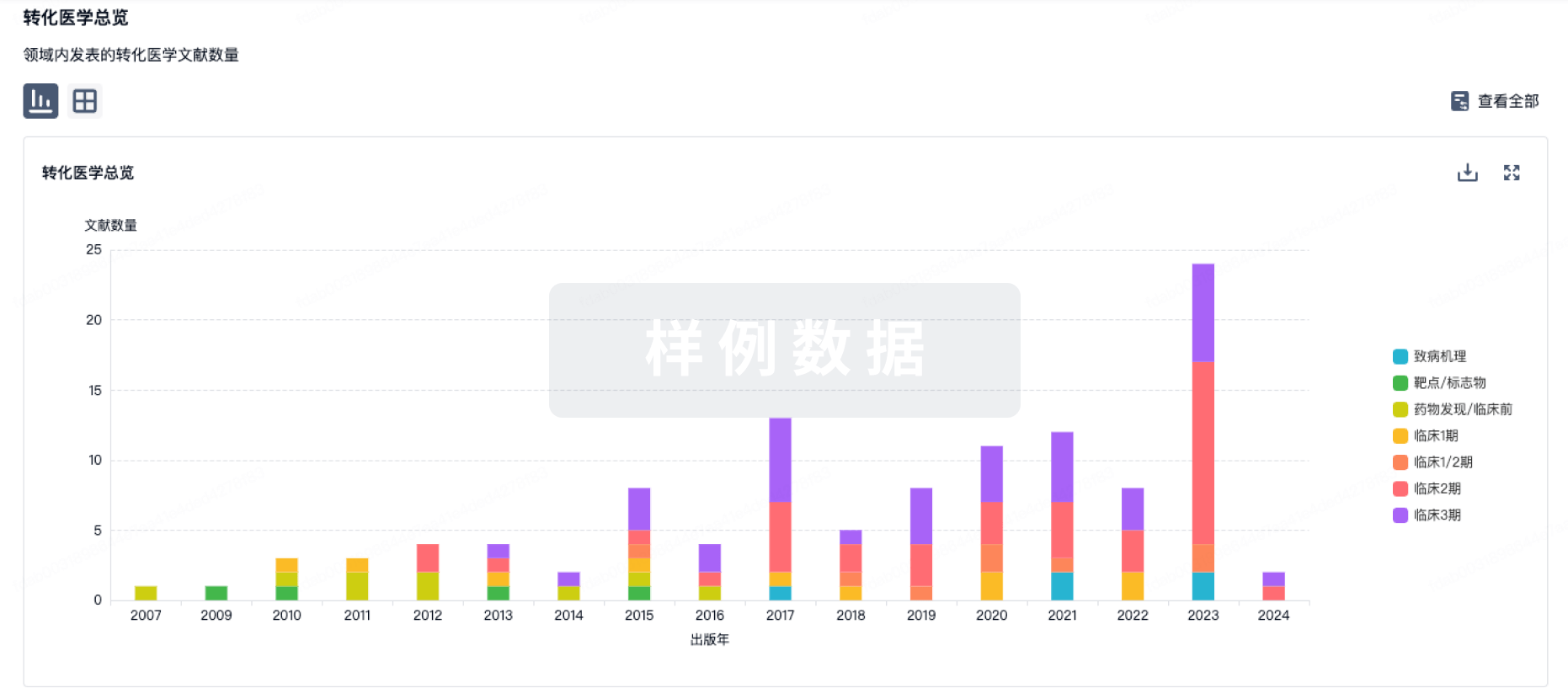
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
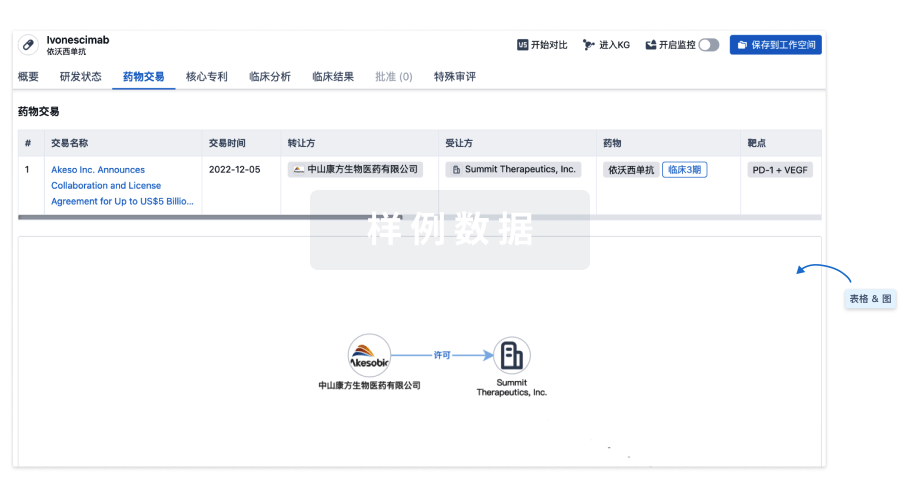
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
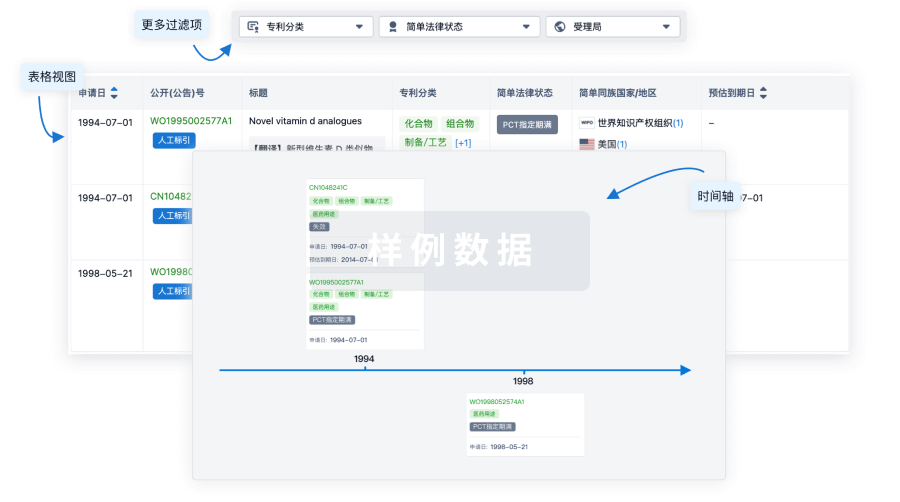
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用