预约演示
更新于:2026-03-22
ZB-004
更新于:2026-03-22
概要
基本信息
原研机构 |
在研机构 |
最高研发阶段临床前 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |


登录后查看时间轴
关联
1
项与 ZB-004 相关的临床试验NCT05794516
A Single Ascending Dose (SAD) Study to Evaluate the Safety and Pharmacokinetics (PK) of ZB004 in Healthy Volunteers
100 项与 ZB-004 相关的临床结果
登录后查看更多信息
100 项与 ZB-004 相关的转化医学
登录后查看更多信息
100 项与 ZB-004 相关的专利(医药)
登录后查看更多信息
5
项与 ZB-004 相关的文献(医药)2007-07-15Toxicology and applied pharmacology3区 · 医学
Cytotoxicity of diacetoxyscirpenol is associated with apoptosis by activation of caspase-8 and interruption of cell cycle progression by down-regulation of cdk4 and cyclin B1 in human Jurkat T cells
3区 · 医学
Article
作者: H PARK ; J KIM ; D JUN ; Y BAE ; W SONG ; Y KIM
To understand the mechanism underlying T-cell toxicity of diacetoxyscirpenol (DAS) from Fusarium sambucinum, its apoptogenic as well as growth retardation activity was investigated in human Jurkat T cells. Exposure to DAS (0.01-0.15 microM) caused apoptotic DNA fragmentation along with caspase-8 activation, Bid cleavage, mitochondrial cytochrome c release, activation of caspase-9 and caspase-3, and PARP degradation, without any alteration in the levels of Fas or FasL. Under these conditions, necrosis was not accompanied. The cytotoxicity of DAS was not blocked by the anti-Fas neutralizing antibody ZB-4. Although the DAS-induced apoptotic events were completely prevented by overexpression of Bcl-xL, the cells overexpressing Bcl-xL were unable to divide in the presence of DAS, resulting from the failure of cell cycle progression possibly due to down-regulation in the protein levels of cdk4 and cyclin B1. The DAS-mediated apoptosis and activation of caspase-8, -9, and -3 were abrogated by either pan-caspase inhibitor (z-VAD-fmk) or caspase-8 inhibitor (z-IETD-fmk). While the DAS-mediated apoptosis and activation of caspase-9 and caspase-3 were slightly suppressed by the mitochondrial permeability transition pore inhibitor (CsA), both caspase-8 activation and Bid cleavage were not affected by CsA. The activated normal peripheral T cells possessed a similar susceptibility to the cytotoxicity of DAS. These results demonstrate that the T-cell toxicity of DAS is attributable to not only apoptosis initiated by caspase-8 activation and subsequent mitochondrion-dependent or -independent activation of caspase cascades, which can be regulated by Bcl-xL, but also interruption of cell cycle progression caused by down-regulation of cdk4 and cyclin B1 proteins.
2003-04-01Cancer gene therapy2区 · 医学
Quantification and characterization of the bystander effect in prostate cancer cells following adenovirus-mediated FasL expression
2区 · 医学
Article
作者: Norris, James S. ; Schwartz, David A. ; Dong, Jian-yun ; Sudarshan, Sunil ; Hyer, Marc L. ; Hannun, Yusuf
Inducing Fas-mediated apoptosis in prostate cancer (PCa) is a promising new therapeutic approach with the potential to overcome delivery issues currently problematic in cancer gene therapy. We have previously demonstrated that a Fas Ligand (FasL) expressing adenovirus (AdGFPFasL(TET)) was able to induce Fas-mediated apoptosis in a panel of PCa cell lines regardless of their Fas-sensitivity as determined by the agonistic Fas antibody CH-11. We now report that AdGFPFasL(TET)-infected cells produce apoptotic bodies and cellular debris that continues to elicit FasL-mediated bystander killing in uninfected neighboring cells. Using light microscopy, we demonstrate that AdGFPFasL(TET)-infected cells release apoptotic bodies and cellular debris into the local environment and that this material will induce bystander killing in Jurkat, PPC-1, and PC-3 target cells, but not in DU145 and K-562 cells. The bystander killing mechanism is mediated through Fas/FasL interaction because it is significantly inhibited if target cells are pretreated with the pan spectrum caspase inhibitor Z-VAD-FMK or the Fas neutralizing antibody ZB-4. Coincubation of PPC-1 target cells with apoptotic bodies and cellular debris (effector material) induce nearly complete target cell killing at a ratio of 1:1 target to effector. Collectively, these data indicate that AdGFPFasL(TET)-infected PCa cells release apoptotic and cellular debris capable of inducing bystander killing in PCa and supports the development of FasL as a gene therapy agent.
1998-09-01European journal of immunology3区 · 医学
Fas (CD95)-transduced signal preferentially stimulates lupus peripheral T lymphocytes
3区 · 医学
Article
作者: Talal, Norman ; Kong, Liping ; Sakata, Atsuko ; Sakata, Kenmei ; Dang, Howard ; Vela-Roch, Norma ; Escalante, Agustin ; Cheng, Jun ; Espinosa, Rolando ; Nakabayashi, Toru
Fas (CD95) is a cell surface receptor whose biological function in circulating peripheral T cells is not well understood. To address the question of abnormal T cell sensitivity to Fas stimulation in systemic lupus erythematosus (SLE), we studied Fas-transduced stimulation and apoptosis in peripheral blood T cells from patients with SLE and normal control. Immobilized anti-Fas monoclonal antibodies (mAb) (imCH-11; IgM type) significantly stimulated SLE T cell proliferation compared to T cells from normal donors and patients with rheumatoid arthritis (p < 0.003 and p < 0.005, respectively). The soluble form of CH-11 and other immobilized anti-Fas mAb (UB-2, ZB-4; IgG type) failed to stimulate lupus T cells while immobilized human Fas ligand did. Furthermore, imCH-11 induced IL-2 and IL-6 mRNA expression. However, imCH-11 activation failed to induce expression of the T cell activation surface molecules CD25 and CD69. Addition of exogenous ceramide, a second messenger for Fas-mediated apoptosis signaling, also induced T cell proliferation in SLE and normal controls. Moreover, fumonisin B1, a specific ceramide synthase inhibitor, and caspase inhibitors markedly suppressed imCH-11 induced T cell proliferation, suggesting that the ceramide pathway may be involved in Fas-transduced stimulation signals in SLE T cells. These results show that SLE T cells have an alteration in the Fas signal transduction pathway leading to cell proliferation. This defect may be important in Fas-mediated peripheral immune homeostasis.

100 项与 ZB-004 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 自身免疫性疾病 | 临床前 | 美国 | 2022-01-30 | |
| 自身免疫性疾病 | 临床前 | 美国 | 2022-01-30 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

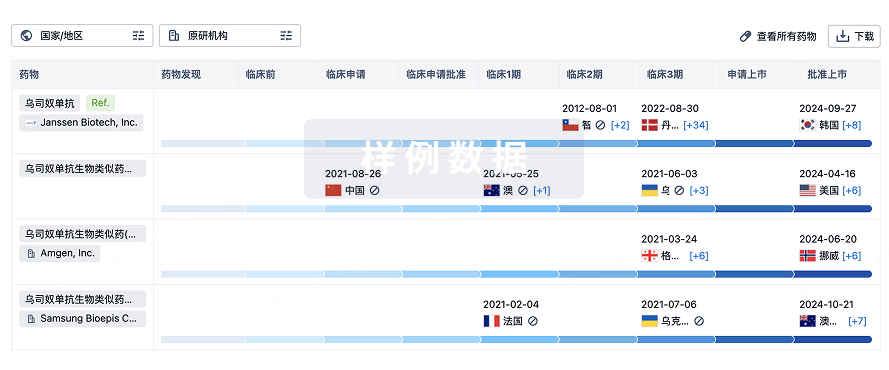
生物类似药
生物类似药在不同国家/地区的竞争态势。请注意临床1/2期并入临床2期,临床2/3期并入临床3期
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或

生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用