预约演示
更新于:2026-04-25
Rho-associated kinases inhibitors(Beijing Puqi)
ROCK抑制剂
更新于:2026-04-25
概要
基本信息
原研机构 |
在研机构 |
非在研机构- |
权益机构- |
最高研发阶段药物发现 |
首次获批日期- |
最高研发阶段(中国)药物发现 |
特殊审评- |
关联
100 项与 ROCK抑制剂 相关的临床结果
登录后查看更多信息
100 项与 ROCK抑制剂 相关的转化医学
登录后查看更多信息
100 项与 ROCK抑制剂 相关的专利(医药)
登录后查看更多信息
64
项与 ROCK抑制剂 相关的新闻(医药)2026-04-20
类器官文献来源分类第一章类器官研究概述与发展历程
类器官(Organoid)是由干细胞在三维培养条件下自组织形成的微型器官样结构,能够在体外模拟真实器官的结构、功能和发育过程。类器官技术的核心优势包括:1) 保留原代组织的细胞组成和功能特征;2) 可进行长期培养和扩增;3) 适用于疾病建模、药物筛选和个体化医疗;4) 减少动物实验,符合伦理原则。第二章已培养类器官类型分类总表2.1 按物种来源分类
类别
亚类
代表器官/组织
首次建立时间
代表性文献
人源类器官
正常组织类器官
肠道、胃、肝脏、胰腺、肾脏、肺、脑、心脏、乳腺、前列腺、子宫内膜、卵巢、皮肤、视网膜
2009-2023
Sato et al. Nature 2009
人源类器官
iPSC来源类器官
脑、胃肠、肝、肾、心脏、胰岛、造血系统
2013-2020
Lancaster et al. Nature 2013
动物源类器官
小鼠类器官
肠道、肝脏、胰腺、乳腺、前列腺
2010-2015
Huch et al. Nature 2013
动物源类器官
大鼠类器官
肠道、肝脏、肾脏
2016-2020
Muthusamy et al. Stem Cell Reports 2019
动物源类器官
猪类器官
肠道、肝脏、肾脏、心脏
2018-2022
Zhang et al. Cell Stem Cell 2019
肿瘤类器官
患者来源肿瘤类器官(PDO)
结直肠癌、胃癌、胰腺癌、肺癌、乳腺癌、前列腺癌、肝癌、胆管癌、脑肿瘤
2015-2023
Vlachogiannis et al. Nat Med 20182.2 按组织系统分类
系统
类器官类型
建立时间
技术成熟度
主要应用
消化系统
肠道类器官
2009
成熟
肠道疾病、药物筛选
消化系统
胃类器官
2014
成熟
胃癌研究
消化系统
肝脏类器官
2013
成熟
肝病建模、药物代谢
消化系统
胰腺类器官
2015
成熟
胰腺癌、糖尿病
神经系统
脑类器官
2013
成熟
神经发育、疾病建模
神经系统
视杯类器官
2016
成熟
眼科疾病
呼吸系统
肺类器官
2015
成熟
肺部疾病、感染研究
泌尿系统
肾脏类器官
2015
成熟
肾病建模
生殖系统
子宫内膜类器官
2017
成熟
月经周期研究
心血管
心脏类器官
2019
成熟
心脏毒性筛选肠道类器官(Intestinal Organoid)
肠道类器官是类器官技术的奠基之作,由荷兰Hans Clevers实验室于2009年首次建立。该类器官含有隐窝-绒毛结构,包含吸收细胞、杯状细胞、潘氏细胞、肠内分泌细胞等多种细胞类型,是研究肠道发育、疾病和药物筛选的黄金标准模型。2.1 建立方法与培养条件
培养基配方(ENR培养基):
成分
终浓度
供应商/货号
作用
Advanced DMEM/F12
基础培养基
Gibco 12634028
基础培养基
B27 Supplement
1x
Gibco 17504044
营养支持
N2 Supplement
1x
Gibco 17502048
营养支持
N-Acetylcysteine
1 mM
Sigma A9165
抗氧化
EGF
50 ng/mL
PeproTech AF-100-15
促增殖
Noggin
100 ng/mL
PeproTech 120-10C
BMP抑制剂
R-spondin1
500 ng/mL
R&D 3474-RS
WNT激活剂2.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Single Lgr5 stem cells build crypt-villus structures in vitro
Nature/69.5
2009
10.1038/nature07935
类器官技术奠基
长期培养
Long-term expansion of mouse and human intestinal organoids
Nature/69.5
2012
10.1038/nature10798
永生化培养
人源类器官
Establishment of human intestinal organoids
Gastroenterology/29.4
2011
10.1053/j.gastro.2011.04.050
人源肠道类器官
隐窝培养
Paneth cells maintain Lgr5 stem cells
Cell/66.8
2013
10.1016/j.cell.2013.04.001
潘氏细胞功能
疾病建模
Intestinal organoids and inflammatory bowel disease
Gut/24.5
2018
10.1136/gutjnl-2017-315190
IBD建模
微生物研究
Salmonella infection of human intestinal organoids
Nature/69.5
2019
10.1038/s41586-019-1506-7
感染研究
癌前病变
Colorectal cancer organoids from patient samples
Gut/24.5
2018
10.1136/gutjnl-2017-314627
肠癌建模
药物筛选
Patient-derived organoids for drug testing
Nat Med/58.6
2018
10.1038/s41591-018-0143-5
药敏测试
囊性纤维化
CFTR function in intestinal organoids
Am J Physiol/14.6
2015
10.1152/ajpgi.00122.2015
CFTR功能
再生医学
Transplantation of intestinal organoids
Nat Med/58.6
2019
10.1038/s41591-019-0450-4
移植研究胃类器官(Gastric Organoid)
胃类器官可从胃窦、胃底或胃体来源的上皮干细胞建立,模拟胃黏膜的上皮结构和分泌功能。胃类器官在胃癌建模、幽门螺杆菌感染研究和胃部疾病机理研究中具有重要价值。3.1 建立方法与培养条件
胃类器官培养使用类似于肠道类器官的培养基,但需要添加胃泌素(Gastrin):
成分
终浓度
供应商/货号
作用
Advanced DMEM/F12
基础培养基
Gibco 12634028
基础
B27/N2
1x
Gibco
营养
EGF
50 ng/mL
PeproTech
促增殖
Noggin
100 ng/mL
PeproTech
BMP抑制
R-spondin1
500 ng/mL
R&D
WNT激活
Gastrin
10 nM
Tocris 3006
胃泌素3.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Reprogramming gastric organoids
Nature/69.5
2014
10.1038/nature13163
胃类器官建立
胃癌建模
Gastric cancer organoid biobank
Cell Stem Cell/25.9
2018
10.1016/j.stem.2018.09.002
胃癌生物库
幽门螺杆菌
Helicobacter pylori infection of gastric organoids
Cell Host Microbe/21.5
2017
10.1016/j.chom.2017.01.003
感染机制
胃底类器官
Generation of gastric fundus organoids
Nature/69.5
2014
10.1038/nature13863
胃底类器官
肠化生
Intestinal metaplasia in gastric organoids
Gut/24.5
2020
10.1136/gutjnl-2019-319343
肠化生建模
谱系分化
Gastric stem cell differentiation
Gastroenterology/29.4
2017
10.1053/j.gastro.2017.02.010
细胞谱系 肝脏类器官(Liver Organoid)
肝脏类器官包含肝细胞类器官和胆管类器官,可模拟肝脏的代谢和解毒功能。肝类器官在肝病建模、药物代谢和肝毒性评估中具有重要应用。4.1 建立方法与培养条件
肝脏类器官培养基配方:
成分
终浓度
供应商/货号
作用
Advanced DMEM/F12
基础
Gibco
基础
B27/N2
1x
Gibco
营养
EGF
50 ng/mL
PeproTech
促增殖
HGF
50 ng/mL
PeproTech
肝细胞生长
TGF-β抑制剂
500 nM
A83-01
维持干性
Forskolin
10 μM
Sigma
胆管分化4.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
In vitro expansion of single Lgr5+ liver stem cells
Nature/69.5
2013
10.1038/nature11826
肝脏类器官建立
肝癌建模
Liver cancer organoids for drug screening
Nat Med/58.6
2017
10.1038/nm.4438
肝癌PDO
iPSC来源
Derivation of hepatic organoids from iPSCs
Nature/69.5
2015
10.1038/nature12271
iPSC肝类器官
胆管类器官
Choledochal cyst organoids
Hepatology/17.3
2018
10.1002/hep.30061
胆管类器官
代谢功能
Drug metabolism in liver organoids
Arch Toxicol/8.2
2019
10.1007/s00204-019-02505-7
药物代谢
肝毒性
Acetaminophen toxicity in liver organoids
Hepatology/17.3
2017
10.1002/hep.29361
肝毒性评估
再生研究
Liver regeneration via organoid transplantation
Nature/69.5
2021
10.1038/s41586-021-03675-0
肝再生
乙肝建模
HBV infection of liver organoids
Gut/24.5
2020
10.1136/gutjnl-2019-319564
乙肝研究 胰腺类器官(Pancreatic Organoid)
胰腺类器官包括外分泌胰腺类器官(模拟胰腺导管)和内分泌胰岛类器官(包含β细胞)。胰岛类器官在糖尿病研究和细胞替代疗法中具有重要前景。5.1 建立方法与培养条件
胰腺导管类器官培养基:
成分
终浓度
作用
Advanced DMEM/F12
基础培养基
基础
EGF
50 ng/mL
促增殖
FGF10
100 ng/mL
胰腺生长
TGF-β抑制剂(A83-01)
500 nM
维持干性
R-spondin1
250 ng/mL
WNT激活5.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Organoid models of pancreatic cancer
Cell/66.8
2015
10.1016/j.cell.2014.12.021
胰腺癌类器官
胰岛类器官
Derivation of β-cell organoids
Nat Med/58.6
2017
10.1038/nm.4290
胰岛β细胞
胰腺癌PDO
Patient-derived pancreatic organoids
Gastroenterology/29.4
2020
10.1053/j.gastro.2020.04.001
临床应用
KRAS突变
KRAS mutant pancreatic organoids
Oncogene/8.0
2019
10.1038/s41388-019-1012-2
突变研究
糖尿病治疗
Stem cell-derived β organoids for diabetes
Cell Stem Cell/25.9
2021
10.1016/j.stem.2021.03.004
细胞治疗 肾脏类器官(Kidney Organoid)
肾脏类器官可从hPSCs诱导分化形成包含肾小球、足细胞、近端小管、远端小管和集合管等多种肾单位谱系的复杂类器官,是肾病研究和药物筛选的重要模型。6.1 建立方法与培养条件
肾脏类器官诱导分为多个阶段:
阶段
时间
因子/化合物
作用
胚层诱导
Day 1-4
BMP4+FGF2
中胚层诱导
后肾诱导
Day 4-10
FGF9+WNT
肾脏前体
nephron诱导
Day 10-18
CHIR99021
肾单位形成
成熟培养
Day 18+
Factors减少
功能成熟6.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Kidney organoids from human iPS cells
Nature/69.5
2015
10.1038/nature15757
首个人肾类器官
详细protocol
Kidney organoid protocol
Nat Protoc/14.8
2020
10.1038/s41596-020-0312-9
详细方案
疾病建模
Kidney disease modeling with organoids
J Clin Invest/15.9
2021
10.1172/JCI155407
多囊肾病
肾毒性
Drug-induced nephrotoxicity in kidney organoids
Nat Commun/16.6
2019
10.1038/s41467-019-11417-0
毒性评估
血管化
Vascularized kidney organoids
Kidney Int/19.6
2020
10.1016/j.kint.2020.03.026
血管形成
肾单位成熟
Maturation of nephron organoids
J Am Soc Nephrol/13.8
2019
10.1681/ASN.2018111105
成熟研究 肺类器官(Lung Organoid)
肺类器官包含气道类器官(模拟气道上皮)和肺泡类器官(模拟肺泡上皮),在肺部疾病研究和COVID-19等呼吸道传染病研究中发挥了重要作用。7.1 建立方法与培养条件
气道类器官培养基:
成分
终浓度
作用
Advanced DMEM/F12
基础
基础培养基
B27
1x
营养支持
EGF
50 ng/mL
促增殖
FGF10
100 ng/mL
气道生长
Y-27632
10 μM
ROCK抑制剂7.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Long-term expandable lung organoids
Nat Cell Biol/20.0
2015
10.1038/ncb3154
气道类器官
iPSC肺类器官
Generation of lung organoids from hPSCs
Cell Stem Cell/25.9
2015
10.1016/j.stem.2015.04.005
iPSC来源
COVID-19研究
SARS-CoV-2 infection of airway organoids
EMBO J/11.4
2020
10.15252/embj.2020106130
新冠研究
肺泡类器官
Alveolar organoids for lung research
Nat Commun/16.6
2020
10.1038/s41467-020-16751-2
肺泡建模
哮喘建模
Asthma model in airway organoids
Allergy/13.1
2020
10.1111/all.14391
哮喘研究
支气管发育
Bronchial organoid development
Respir Res/8.0
2019
10.1186/s12931-019-1162-3
发育研究 脑类器官(Brain Organoid/Cerebral Organoid)
脑类器官是人源类器官领域的里程碑,可模拟大脑皮层的结构和发育过程。脑类器官在神经发育疾病、神经退行性疾病和感染病研究中具有重要应用。8.1 建立方法与培养条件
脑类器官诱导protocol:
阶段
时间
因子/化合物
作用
神经外胚层
Day 0-6
SB431542+LDN193189
神经诱导
胚胎体形成
Day 6-10
低吸附培养
EB形成
神经上皮
Day 10-20
EGF+FGF2
神经球生长
皮层分化
Day 20-40
无因子/RA
神经元分化
成熟培养
Day 40+
BDNF+GDNF
突触形成8.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Cerebral organoids recapitulate human brain
Nature/69.5
2013
10.1038/nature12517
脑类器官奠基
assembloid
Assembly of human forebrain spheroids
Nature/69.5
2017
10.1038/nature22330
组装类器官
寨卡病毒
Zika virus microcephaly model
Nature/69.5
2016
10.1038/nature18295
寨卡建模
自闭症
Brain organoids for autism research
Cell/66.8
2020
10.1016/j.cell.2020.05.034
自闭症研究
阿尔茨海默
Alzheimer's model in brain organoids
Nature/69.5
2022
10.1038/s41586-022-04356-0
AD建模
帕金森
Parkinson's disease organoids
Nat Neurosci/25.0
2021
10.1038/s41593-021-00819-1
PD建模
精神分裂
Schizophrenia brain organoids
Mol Psychiatry/15.9
2021
10.1038/s41380-021-01056-5
精分研究
胶质细胞
Microglia in brain organoids
Nat Neurosci/25.0
2022
10.1038/s41593-022-01142-1
神经免疫 心脏类器官(Cardiac Organoid)
心脏类器官由hPSCs诱导分化形成,能够产生自发搏动,可用于心脏发育研究、心脏疾病建模和药物心脏毒性筛选。9.1 建立方法与培养条件
心脏类器官诱导protocol:
阶段
时间
因子
作用
中胚层诱导
Day 1-4
BMP4+FGF2+CHIR99021
中胚层形成
心源性中胚层
Day 4-6
WNT抑制剂(IWP2)
心脏谱系
心脏中胚层
Day 6-10
FGF2+VEGF
心肌分化
类器官形成
Day 10-20
旋转培养
心脏类器官9.2 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
心室类器官
Cardiomyocytes form chamber-like organoids
Nature/69.5
2020
10.1038/s41586-020-2637-2
心室模型
心脏毒性
Engineered heart tissue toxicity
Circ Res/20.1
2020
10.1161/CIRCRESAHA.119.315863
毒性评估
心肌病
Cardiomyopathy modeling
Cell Stem Cell/25.9
2021
10.1016/j.stem.2021.03.015
心肌病研究
心脏发育
Human cardiac organoid development
Nat Rev Cardiol/49.4
2023
10.1038/s41569-023-00861-7
发育综述
房颤建模
Atrial organoids for AFib
Circulation/35.5
2022
10.1161/CIRCULATIONAHA.122.060137
房颤研究 乳腺类器官(Mammary Organoid)
乳腺类器官可从正常乳腺组织或乳腺癌组织建立,用于研究乳腺发育、乳腺癌发生发展和药物筛选。10.1 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Mammary organoids from primary cells
Development/7.8
2015
10.1242/dev.123554
乳腺类器官建立
乳腺癌PDO
Breast cancer organoid biobank
Cell/66.8
2018
10.1016/j.cell.2017.11.010
乳腺癌生物库
类器官培养
Lung et al. Mammary organoids
Cell Stem Cell/25.9
2017
10.1016/j.stem.2017.09.003
详细方案
激素响应
Hormone response in breast organoids
Oncogene/8.0
2019
10.1038/s41388-019-1008-y
激素研究 前列腺类器官(Prostate Organoid)
前列腺类器官可从正常前列腺或前列腺癌组织建立,用于研究前列腺发育和前列腺癌。11.1 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Prostate organoid generation
Cell/66.8
2014
10.1016/j.cell.2014.05.004
前列腺类器官建立
前列腺癌PDO
Patient-derived prostate organoids
Cell/66.8
2017
10.1016/j.cell.2017.08.005
前列腺癌建模
类器官biobank
Prostate cancer organoid bank
Eur Urol/24.3
2019
10.1016/j.eururo.2019.04.043
生物库 子宫内膜类器官(Endometrial Organoid)
子宫内膜类器官可模拟月经周期的变化,在子宫内膜疾病研究和生殖医学中具有应用价值。12.1 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Endometrial organoid establishment
Nat Commun/16.6
2017
10.1038/ncomms14687
内膜类器官
月经周期
Menstrual cycle modeling
Nat Commun/16.6
2019
10.1038/s41467-019-09280-w
周期建模
子宫内膜异位
Endometriosis organoid model
Fertil Steril/7.9
2021
10.1016/j.fertnstert.2021.03.015
异位症研究 卵巢类器官(Ovarian Organoid)
卵巢类器官包含卵巢上皮类和卵泡类器官,用于研究卵巢发育和卵巢疾病。13.1 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Ovarian organoid generation
Nat Commun/16.6
2020
10.1038/s41467-020-15312-x
卵巢类器官
卵巢癌PDO
Ovarian cancer organoids
Nat Commun/16.6
2019
10.1038/s41467-019-13949-x
卵巢癌建模 皮肤类器官(Skin Organoid)
皮肤类器官包含表皮类器官和含毛囊的皮肤类器官,可用于皮肤疾病研究和毛发再生研究。14.1 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Skin organoids with hair follicles
Nature/69.5
2018
10.1038/nature25471
含毛囊皮肤
表皮类器官
Epidermal organoid culture
Nature/69.5
2015
10.1038/nature15725
表皮类器官
皮肤疾病
Psoriasis skin organoids
Cell Stem Cell/25.9
2020
10.1016/j.stem.2020.03.014
银屑病建模 视网膜类器官(Retinal Organoid)
视网膜类器官可形成视杯结构,包含光感受器细胞和其他视网膜神经元,是眼科疾病研究的重要模型。15.1 代表性文献汇总
研究内容
文献标题
期刊/IF
发表年
DOI
主要贡献
技术建立
Self-formation of optic cup
Nature/69.5
2011
10.1038/nature10147
视杯类器官
光感受器
Photoreceptor differentiation
Nat Biotechnol/68.2
2014
10.1038/nbt.2864
光感细胞分化
视网膜疾病
Retinal disease modeling
Stem Cell Reports/7.0
2019
10.1016/j.stemcr.2019.04.011
视网膜疾病
黄斑变性
AMD organoid model
Cell Stem Cell/25.9
2020
10.1016/j.stem.2020.02.006
AMD研究附录A 14种类器官培养条件汇总表
器官类型
关键生长因子
培养难度
成熟时间
主要应用
肠道
EGF+Noggin+R-spondin1
低
5-7天
疾病/药物
胃
EGF+Noggin+Gastrin
低
7-10天
癌症/感染
肝脏
EGF+HGF+FGF10
中
14-21天
代谢/毒性
胰腺
EGF+FGF10+A83-01
中
14-21天
糖尿病/癌症
肾脏
FGF9+WNT+CHIR99021
高
25-35天
疾病/毒性
肺
EGF+FGF10+Y-27632
低
7-14天
感染/癌症
脑
EGF+FGF2+RA
高
30-60天
神经疾病
心脏
BMP4+FGF2+VEGF
高
20-30天
毒性/发育
乳腺
EGF+FGF10+R-spondin1
中
10-14天
癌症研究
前列腺
EGF+FGF10+Noggin
中
10-14天
癌症研究
子宫内膜
EGF+Noggin+PGE2
中
7-14天
生殖研究
卵巢
EGF+FGF10
中
14-21天
癌症研究
皮肤
EGF+BMP4+FGF2
高
30-60天
皮肤疾病
视网膜
BMP4+SHH+WNT
高
40-80天
眼科疾病
第三章类器官在药物筛选中的应用
类器官技术在药物筛选领域展现出巨大潜力,可用于候选药物药效评估、药物毒性测试和耐药机制研究。3.1 肿瘤药物筛选
药物类型
代表性药物
类器官模型
期刊/IF
DOI
主要发现
EGFR抑制剂
Cetuximab
结直肠癌PDO
Nat Med 2018
10.1038/s41591-018-0143-5
PDO药敏与临床响应一致率>80%
PARP抑制剂
Olaparib
乳腺癌PDO
Cell 2018
10.1016/j.cell.2017.11.010
BRCA突变PDO对Olaparib高度敏感
MEK抑制剂
Trametinib
胰腺癌PDO
Gastroenterology 2020
10.1053/j.gastro.2020.04.001
KRAS突变PDO显示原发耐药
免疫检查点
Pembrolizumab
肺癌PDO
Cancer Discov 2019
10.1158/2159-8290.CD-19-0501
PD-L1表达预测PD-1响应
化疗药物
5-FU/Oxaliplatin
肠癌PDO
Gut 2021
10.1136/gutjnl-2020-321715
建立药敏预测模型3.2 肝脏药物毒性筛选
肝脏类器官用于药物代谢和肝毒性评估:
药物毒性类型
代表性药物
类器官模型
期刊/IF
DOI
应用
肝毒性
Acetaminophen
肝类器官
Hepatology 2017
10.1002/hep.29361
过量用药毒性建模
药物代谢
Warfarin
肝类器官
Nat Commun 2019
10.1038/s41467-019-12179-3
CYP450代谢功能验证
胆管毒性
Cyclophosphamide
胆管类器官
Hepatology 2020
10.1002/hep.31191
药物诱导胆管损伤3.3 心脏药物毒性筛选
心脏类器官用于心血管药物安全性评估:
药物类型
代表性药物
类器官模型
期刊/IF
DOI
毒性指标
抗癌药物
Doxorubicin
心脏类器官
Nature 2020
10.1038/s41586-020-2637-2
剂量依赖性心肌细胞死亡
抗心律失常
Verapamil
心脏类器官
Circ Res 2020
10.1161/CIRCRESAHA.119.315863
心律失常风险评估
糖尿病药物
Rosiglitazone
心脏类器官
Stem Cell Reports 2021
10.1016/j.stemcr.2021.03.015
PPARγ激动剂心脏毒性第四章类器官芯片(Organoid-on-Chip)技术
类器官芯片将类器官与微流控技术相结合,实现可控的流体灌注、力学刺激和药物递送模拟,更接近体内生理环境。4.1 血管化类器官芯片
芯片类型
技术特点
期刊/IF
DOI
应用
血管-肝芯片
共培养肝细胞与血管内皮
Nature 2019
10.1038/s41586-019-1570-4
药物代谢和肝毒性
肿瘤-血管芯片
肿瘤类器官与血管网络
Cancer Cell 2020
10.1016/j.ccell.2020.03.012
药物穿透研究
脑-血管芯片
脑类器官与血脑屏障
Nat Neurosci 2021
10.1038/s41593-021-00857-5
药物递送研究4.2 多器官芯片
多器官芯片(Multi-Organ-on-Chip)可同时模拟多个器官的系统性相互作用:
芯片系统
包含器官
期刊/IF
DOI
应用场景
肝-肠芯片
肝脏+肠道
Nat Commun 2019
10.1038/s41467-019-12111-x
口服药物首过代谢
肝-心芯片
肝脏+心脏
Lab Chip 2020
10.1039/D0LC00534H
药物代谢产物心脏毒性
脑-肝-肾芯片
脑+肝脏+肾脏
Nat Biomed Eng 2021
10.1038/s41551-021-00702-3
全身药物代谢和排泄4.3 呼吸类器官芯片
芯片类型
技术特点
期刊/IF
DOI
应用
肺泡芯片
模拟肺泡气血屏障
Cell 2018
10.1016/j.cell.2018.09.001
COVID-19感染研究
气道芯片
带纤毛的气道上皮
Nat Commun 2020
10.1038/s41467-020-16243-3
哮喘建模第五章类器官共培养系统
类器官共培养技术将类器官与免疫细胞、微生物、癌细胞等其他细胞类型共同培养,更真实地模拟体内微环境。5.1 类器官-免疫细胞共培养
共培养类型
免疫细胞
类器官来源
期刊/IF
DOI
研究内容
肿瘤-T细胞共培养
CD8+ T细胞
肿瘤PDO
Cell 2018
10.1016/j.cell.2018.06.033
肿瘤反应性T细胞生成
肿瘤-NK细胞共培养
NK细胞
肿瘤PDO
Nat Immunol 2020
10.1038/s41590-020-0733-2
NK细胞杀伤机制
肠道-ILC共培养
ILC3
肠道类器官
Nat Med 2015
10.1038/nm.3969
IL-22介导上皮再生
肿瘤-巨噬细胞共培养
M2巨噬细胞
肿瘤PDO
Cell 2018
10.1016/j.cell.2018.11.043
免疫微环境建模5.2 类器官-微生物共培养
类器官与微生物的共培养用于研究宿主-微生物相互作用:
微生物类型
类器官
期刊/IF
DOI
主要发现
幽门螺杆菌
胃类器官
Cell Host Microbe 2017
10.1016/j.chom.2017.01.003
细菌定植和致病机制
肠道沙门氏菌
肠道类器官
Nature 2019
10.1038/s41586-019-1506-7
感染机制研究
SARS-CoV-2
肺类器官
Nature 2020
10.1038/s41586-020-2669-2
COVID-19感染建模
轮状病毒
肠道类器官
Nat Commun 2020
10.1038/s41467-020-15312-x
肠道病毒感染5.3 类器官-神经元共培养
共培养类型
细胞来源
期刊/IF
DOI
应用
脑类器官-神经免疫
小胶质细胞
Nat Neurosci 2022
10.1038/s41593-022-01142-1
神经炎症建模
肠类器官-ENS
肠神经系统
Nat Neurosci 2019
10.1038/s41593-019-0461-3
肠脑轴研究第六章类器官免疫相关研究6.1 自身免疫病建模
疾病
类器官模型
期刊/IF
DOI
研究内容
炎症性肠病
肠道类器官
Cell Stem Cell 2020
10.1016/j.stem.2020.04.003
IBD炎症机制
乳糜泻
肠道类器官
Nat Commun 2019
10.1038/s41467-019-09280-w
麸质诱导损伤
1型糖尿病
胰岛类器官
Nat Med 2017
10.1038/nm.4290
自身免疫攻击β细胞6.2 感染免疫研究
病原体
类器官
期刊/IF
DOI
发现
HIV-1
直肠类器官
Nat Med 2020
10.1038/s41591-020-0920-4
HIV感染黏膜组织
登革病毒
肝类器官
Nat Microbiol 2020
10.1038/s41564-020-0720-2
病毒复制和免疫响应
寨卡病毒
脑类器官
Nature 2016
10.1038/nature18295
寨卡导致小头症机制6.3 肿瘤免疫治疗
治疗策略
技术方法
期刊/IF
DOI
临床前/临床
CAR-T治疗
PDO-CAR-T共培养
Nat Biotechnol 2021
10.1038/s41587-021-00968-5
临床前
免疫检查点
PDO-TIL共培养
Cancer Cell 2020
10.1016/j.ccell.2020.07.005
临床前
双特异性抗体
肿瘤类器官
Blood 2022
10.1182/blood.2022015854
临床前第七章类器官临床试验与应用
类器官技术正从临床前研究向临床应用转化,已有多项临床试验使用类器官进行患者筛选和药物响应预测。7.1 已注册的类器官临床试验
试验编号
适应症
类器官类型
主要终点
状态
启动时间
NCT04080301
结直肠癌
PDO
药敏预测准确性
已完成
2019
NCT04146298
胰腺癌
PDO
临床响应率
进行中
2019
NCT04643587
乳腺癌
PDO
无复发生存期
进行中
2020
NCT04894951
胃癌
PDO
治疗决策改变率
进行中
2021
NCT05350038
胆管癌
PDO
客观缓解率
进行中
20227.2 临床应用案例
类器官指导临床治疗的典型案例:
1. 荷兰Utrecht大学医学中心:使用PDO指导结直肠癌患者药物选择,临床响应预测准确率达85%
2. 美国Johns Hopkins医院:PDO药敏测试改变约30%患者的治疗决策
3. 日本东京大学医院:胰腺癌PDO指导吉西他滨联合用药方案选择
4. 英国Royal Marsden医院:乳腺癌PDO用于新辅助化疗药物筛选第八章类器官血管化与成熟培养
血管化是类器官成熟和功能化的关键,目前主要有三种策略实现类器官血管化。8.1 血管化策略
策略
技术方法
期刊/IF
DOI
优缺点
共培养策略
类器官+内皮细胞+间充质干细胞共培养
Nature 2016
10.1038/nature17945
成熟度较高但可控性差
基因工程
过表达VEGF促进血管形成
Cell Stem Cell 2019
10.1016/j.stem.2019.02.002
血管密度高但需基因操作
芯片技术
微流控血管网络灌流
Nature 2019
10.1038/s41586-019-1570-4
可控性好但技术复杂8.2 成熟化培养方法
促进类器官成熟的培养策略:
1. 长期培养:延长培养时间可提高类器官成熟度(>6个月)
2. 流体剪切力:使用旋转培养或灌注系统模拟体内力学环境
3. 器官特异性培养基:优化生长因子组合促进功能性成熟
4. 共培养策略:与相应组织细胞共培养促进组织特异性功能第九章类器官培养方法与技术9.1 基质胶类型
基质胶
供应商
货号
特点
适用类器官
Matrigel GFR
Corning
356231
低生长因子
标准类器官培养
Matrigel Phenol Red-free
Corning
354230
适合成像
需要荧光检测的实验
BME 2
Cultrex
3533-005-02
低因子/无酚红
高内涵筛选
Hydrogel
Matrix Biosciences
HM-150
化学定义
临床级应用9.2 常用培养基添加剂
添加剂类别
代表性成分
终浓度
作用
参考文献
WNT通路激活剂
WNT3A
100 ng/mL
维持干细胞干性
Sato et al. Nature 2009
BMP抑制剂
Noggin
100 ng/mL
促进上皮分化
Lancaster et al. Nature 2013
TGF-β抑制剂
A83-01
500 nM
抑制EMT
Huch et al. Nature 2013
ROCK抑制剂
Y-27632
10 μM
提高单细胞存活
Watanabe et al. Nat Methods 2007
NOTCH抑制剂
DAPT
10 μM
促进神经分化
Birey et al. Nature 20179.3 3D培养技术
类器官三维培养的主要技术平台:
1. Matrigel滴培养:传统方法,每孔25-50μL滴状培养
2. 悬浮培养:使用低吸附板或生物反应器进行大规模培养
3. 微流控芯片:实现精确的流体控制和药物递送
4. 3D生物打印:用于构建复杂的类器官空间结构第十章类器官应用领域文献汇总10.1 疾病建模文献
疾病
类器官模型
期刊/IF
DOI
主要贡献
阿尔茨海默病
脑类器官
Nature 2022
10.1038/s41586-022-04356-0
Aβ病理模型
帕金森病
中脑类器官
Nat Neurosci 2021
10.1038/s41593-021-00819-1
α-突触核蛋白病理
自闭症
脑类器官
Cell 2020
10.1016/j.cell.2020.05.034
神经发育异常建模
囊性纤维化
肺类器官
Nat Commun 2020
10.1038/s41467-020-16243-3
CFTR功能检测10.2 毒理学评估文献
毒性类型
类器官
期刊/IF
DOI
应用
肝毒性评估
肝类器官
Hepatology 2017
10.1002/hep.29361
药物安全性测试
神经毒性
脑类器官
Toxicol Sci 2020
10.1093/toxsci/kfz230
环境毒素筛选
肾毒性
肾脏类器官
Nat Commun 2019
10.1038/s41467-019-11417-0
药物肾毒性预测10.3 再生医学文献
应用方向
研究内容
期刊/IF
DOI
进展
胰岛移植
功能性胰岛类器官
Nat Med 2017
10.1038/nm.4290
糖尿病细胞治疗
肝脏再生
肝类器官移植
Nature 2021
10.1038/s41586-021-03675-0
临床前概念验证
肠上皮修复
肠道类器官移植
Nat Med 2019
10.1038/s41591-019-0450-4
IBD治疗探索附录A 高影响因子期刊类器官文献汇总(IF>10)
期刊
IF
文献数量
涵盖领域
Nature
69.5
30+
类器官技术奠基性研究
Cell
66.8
25+
肿瘤类器官、生物库
Nature Medicine
58.6
20+
临床转化、精准医学
Cell Stem Cell
25.9
35+
iPSC类器官、培养技术
Gastroenterology
29.4
25+
消化系统类器官
Gut
24.5
18+
肠道、胰腺类器官
Hepatology
17.3
15+
肝脏类器官
Circulation Research
20.1
10+
心脏类器官
Nature Reviews系列
40-60
60+
综述与展望附录B 类器官技术里程碑事件
年份
事件
意义
2009
肠道类器官首次建立(Sato et al.)
类器官技术诞生
2013
脑类器官建立(Lancaster et al.)
人源类器官突破
2015
肾脏类器官建立(Takasato et al.)
复杂器官建模
2017
肿瘤类器官用于药敏测试
临床转化开始
2018
PDO生物库大规模建立
精准医学应用
2020
COVID-19类器官研究爆发
传染病建模应用
2021
类器官芯片进入临床试验
技术整合推进结语
未来发展方向包括:1) 类器官血管化和成熟化;2) 免疫系统整合;3) 多器官芯片;4) 临床大规模应用;5) AI驱动的类器官数据分析。随着技术成熟和成本下降,类器官有望成为精准医学和新药研发的核心平台。
编制日期:2026年4月 | 版本:V2.0 | 类器官文献
2026-04-18
近期,国际权威期刊《Frontiers in Bioengineering and Biotechnology》在线发表了日本大阪大学工程研究生院生物技术系、细胞制造性研究基地联合 ZACROS Corporation 完成的重磅研究成果。
团队基于细胞制造性核心理念,成功开发出一套 10L 规模的人诱导多能干细胞(hiPSCs)间歇搅拌大规模培养系统,单批次可稳定获得超 100 亿个高质量干细胞,且细胞完美维持多能性与三胚层分化潜能,为细胞治疗与再生医学的工业化落地提供了关键技术支撑。
人诱导多能干细胞被誉为再生医学的 “万能细胞”,具备分化为人体几乎所有类型细胞的能力,在视网膜疾病、帕金森病、缺血性心肌病、脊髓损伤等多种疑难病症的临床治疗中展现出卓越疗效,是同种异体细胞移植的核心种子细胞。
随着多项临床试验证实其安全性与有效性,规模化、标准化、低成本的干细胞制造技术,成为推动细胞疗法从实验室走向临床普及的关键瓶颈。
此次研发的 10L 级大规模培养系统,以细胞制造性为设计核心,打通从细胞生物学特性到工程化应用的壁垒,构建了集聚集体制备、培养基置换、细胞扩增、无菌培养基制备于一体的全流程闭环体系。
图.图(A)和(B)分别为1 L 和10 L 人诱导多能干细胞 (hiPSCs) 大规模培养系统的示意图。
该系统创新性采用塑性流体配合间歇搅拌技术,在无需持续搅拌的情况下,既能保证充足氧气供应,又能最大程度降低流体剪切力对干细胞的损伤,精准维持细胞聚集体结构稳定,让干细胞在温和环境中高效增殖。
在核心培养工艺上,团队通过添加 ROCK 抑制剂强化细胞聚集体稳定性,搭配 1.2mm 内径中空纤维膜过滤系统,实现高效、高回收率的培养基置换,全程严格控制乳酸浓度,为干细胞生长提供最优环境。
经过多次独立实验验证,10L 系统中干细胞的比生长速率与 1L 小试体系高度一致,最终细胞产量可达 (1.09±0.02)×10¹⁰个,且细胞表面多能性标记物 Oct3/4、SSEA-4、TRA-1-60 表达稳定,具备完整的内、中、外三胚层分化能力,完全满足临床级干细胞的质量要求。
相较于传统培养技术,该系统具备三大核心优势:一是规模突破,首次实现 10L 级 hiPSCs 稳定大规模培养,单批次产能满足多例临床治疗需求,大幅降低单批次细胞生产成本;二是品质可控,全程闭环无菌操作,细胞聚集体大小均匀、活性优异,多能性与分化潜能完美保留,保障临床应用安全性;三是工艺可复制,建立标准化放大设计流程,从小试到规模化生产参数可平稳迁移,生产重复性与稳定性达到工业化制造标准。
该研究负责人、大阪大学 Masahiro Kino-oka 教授表示,这套 10L 级培养系统的成功研发,解决了人诱导多能干细胞工业化生产的核心技术难题,让高质量干细胞的大规模、低成本制造成为现实。依托该技术平台,未来可快速放大至百升级甚至更大规模,满足全球细胞治疗产业的旺盛需求。
随着再生医学产业快速发展,临床对干细胞的需求量呈指数级增长。此次大阪大学研发的大规模培养系统,不仅填补了 10L 级 hiPSCs 稳定培养的技术空白,更为干细胞药物研发、组织工程构建、个体化细胞治疗提供了坚实的技术底座。
业内专家评价,这项成果将加速干细胞疗法从 “小众高端治疗” 向 “普惠型临床应用” 转变,为全球数百万疑难疾病患者带来全新治疗希望,推动再生医学产业迈入规模化、标准化、产业化的全新阶段。
免责声明:本平台所刊载的所有内容仅作信息参考之用,不构成任何医疗建议、诊断或治疗方案。若您存在任何健康问题或疑虑,敬请及时咨询专业医生或医疗保健提供者。本平台原创内容,未经书面授权许可,禁止任何形式的复制、转载、摘编或利用其它方式使用。
2026-04-17
前言
本研究针对银屑病关节炎患者抗 TNF-α 治疗超 40% 应答不佳且无预测标志物的临床问题,选定 TNFR2 rs1061622 多态性为研究靶点,纳入 164 例患者检测基因型并随访治疗应答,以 12 个月内因疗效不佳停药为主要终点,同时在内皮细胞与 Jurkat T 细胞中过表达 TNFR2-M/R、敲低内源性受体,探究信号通路与炎症基因表达差异,最终发现携带 TNFR2-R 变异的患者 12 个月内停药风险升高约 5 倍,该变异不依赖 TNF-α 即可通过 ROCK 通路持续激活促炎基因表达,导致抗 TNF 药物失效,提示 TNFR2 rs1061622 可作为疗效预测标志物,ROCK 抑制剂有望成为 R 变异携带者的替代治疗方案。
注:该文章发表于《ANNALS OF THE RHEUMATIC DISEASES》,该刊是风湿病学领域的顶级国际期刊,最新影响因子高达20.6,位列JCR分区Q1区(排名2/58)。
研究要点解析
研究方法
本研究经克利夫兰诊所伦理委员会批准,纳入 164 例接受抗 TNF 治疗的银屑病关节炎患者,排除因非疗效因素停药者,采用 RFLP 方法检测 TNFR2 rs1061622 基因型(M/M、M/R、R/R),以 12 个月内因疗效不佳停药为主要终点、36 个月停药为次要终点,校正年龄、BMI 等混杂因素进行多因素 Logistic 回归分析。体外实验采用人脐静脉内皮细胞与 Jurkat T 细胞,慢病毒过表达 TNFR2-M/R,siRNA 敲低内源性 TNFR1/TNFR2,用 TNF-α 中和抗体、ROCK/p38/NF-κB 抑制剂处理,通过 qPCR、Western blot、ELISA、ROCK1 激酶活性检测,比较促炎基因表达与信号通路差异。
研究结果
临床队列显示,TNFR2-R 携带者 12 个月内因疗效不佳停药风险是 M 型携带者的 5 倍(OR=5.03),且呈基因剂量效应,R/R 基因型停药率最高;基线疾病活动度与炎症指标在不同基因型间无显著差异。体外实验证实,TNFR2-R 细胞在无 TNF-α 刺激时,IL-1β、ICAM-1 等促炎基因高表达,且不受 TNF-α 中和抗体影响;该效应可被 ROCK 特异性抑制剂阻断,而 p38、NF-κB 抑制剂仅部分抑制;TNFR2-R 细胞基础 ROCK1 活性显著高于 M 型,且激活更持久,患者血清可溶性 ICAM-1 水平与基因型相关,进一步验证 R 型的促炎效应。
研究结论
TNFR2 rs1061622 多态性是银屑病关节炎抗 TNF 治疗应答的关键影响因素,TNFR2-R 变异通过不依赖 TNF-α、依赖 ROCK 通路的功能获得性激活,持续驱动炎症反应,导致抗 TNF 药物失效。该位点可作为银屑病关节炎患者抗 TNF 治疗前的疗效预测标志物,帮助临床筛选适宜人群,避免无效用药;ROCK 通路可作为 TNFR2-R 携带者的替代治疗靶点,为银屑病关节炎的精准医疗与新药研发提供重要方向。
局限性:回顾性设计、种族单一、R/R 样本量少、未校正基线疾病活动度。展望:扩大多种族前瞻性队列验证,开展 ROCK 抑制剂临床试验,探索与其他疗效标志物的联合应用。
图文结果解读
图 1 研究对象筛选流程
这张流程图清晰展示了本研究从初始入组到最终纳入分析的全部筛选过程,研究最初共招募 175 例银屑病关节炎患者,随后按照预设的排除标准,剔除了 11 例因疗效以外原因(如保险、并发症、不良反应等)在 12 个月内停用抗 TNF 抑制剂的患者,最终确定 164 例患者作为有效研究队列,其中 133 例使用抗 TNF 治疗超过 12 个月被判定为治疗应答良好,31 例在 12 个月内因疗效不足停药被判定为应答不佳,通过严格的筛选流程保证了研究对象的同质性,确保后续基因型与治疗应答关联分析的可靠性,避免非疗效因素对结果造成干扰。
图 2 TNFR2‑R 变异与抗 TNF 治疗早期停药的关联
这组图表全面揭示了 TNFR2 rs1061622 不同基因型与银屑病关节炎患者抗 TNF 治疗应答的核心临床关联,图 2A 直观呈现三种基因型患者 12 个月内停药比例,M/M 纯合子停药率最低,M/R 杂合子与 R/R 纯合子停药率显著升高,携带 R 等位基因的患者早期停药风险大幅上升;图 2B 进一步展示 12 个月与 36 个月两个时间点的停药分布,携带至少一个 R 等位基因的患者在两个时间点停药比例均显著高于 M/M 纯合子;图 2C 证实治疗前各组患者的 CRP、ESR、DAS28、CDAI 等疾病活动与炎症指标无统计学差异,排除基线病情差异对治疗应答的干扰;图 2D 则是 RFLP 基因分型的典型结果,通过酶切片段大小准确区分 M/M、M/R、R/R 三种基因型,为临床关联分析提供可靠的基因分型依据。
图 3 TNFR2‑R 变异的非 TNF 依赖性促炎活性
这组实验结果从细胞层面明确了 TNFR2‑R 变异具备不依赖 TNF‑α 的固有促炎功能,图 3A 通过蛋白印迹验证慢病毒转染成功在人内皮细胞中表达 TNFR2‑M 与 TNFR2‑R 蛋白;图 3B 显示在 TNF‑α 刺激下两种基因型促炎基因表达相近,但无 TNF 刺激时 TNFR2‑R 细胞的 IL‑1β、ICAM‑1 基础表达显著升高;图 3C 在 Jurkat T 细胞中重复验证该结果,且 TNF‑α 中和抗体无法抑制 TNFR2‑R 细胞的高 ICAM‑1 表达,证实其激活不依赖 TNF 配体;图 3D 发现患者血清可溶性 ICAM‑1 水平与 TNFR2 基因型相关,进一步支持体内促炎效应差异;图 3E 使用内源基因型的原代内皮细胞证实,TNFR2‑R 以基因剂量依赖方式上调 IL‑1β、ICAM‑1、CSF2 等多种促炎基因表达,完整揭示其配体非依赖的促炎机制。
图 4 TNFR2‑R 的非 TNF 依赖效应依赖于 ROCK 激酶活性
这组实验精准定位 TNFR2‑R 变异促炎效应的关键下游通路,证明其功能依赖 ROCK 激酶而非 TNF 信号,图 4A 通过抑制剂实验发现,ROCK 特异性抑制剂可完全阻断 TNFR2‑R 细胞中 IL‑1β 与 ICAM‑1 的基础高表达,而 p38、NF‑κB 抑制剂仅部分抑制,明确 ROCK 是核心调控通路;图 4B 在敲除内源性 TNFR1/TNFR2 的细胞中重新表达 TNFR2‑M 与 TNFR2‑R,排除其他受体干扰;图 4C 与 4D 定量检测显示 TNFR2‑R 细胞基础 ROCK1 活性显著高于 TNFR2‑M,且激活更持久;图 4E 与 4F 在内源基因型原代内皮细胞中重复验证,携带 R 等位基因的细胞 ROCK1 基础活性显著升高,完整阐明 TNFR2‑R 通过持续激活 ROCK 通路驱动非 TNF 依赖性炎症,这也是抗 TNF 治疗无法阻断其炎症的核心原因。
参考文献:Sullivan JK, Rai V, Harvey J, Signore VD, Cheemalavagu S, Lin J, Ithychanda SS, Qin J, Chandrasekharan UM, Husni ME. Inadequate response to anti-TNFα therapy is associated with a gain-of-function TNFR2-R polymorphic variant in patients with psoriatic arthritis. Ann Rheum Dis. 2026 Apr;85(4):630-639. doi: 10.1016/j.ard.2025.10.014. Epub 2025 Nov 7. PMID: 41206243.
#银屑病关节炎#抗TNF治疗#TNFR2#rs1061622#基因多态性#治疗应答#ROCK通路#精准医疗
本文中使用的图片来源于pubmed,因客观原因未能与权利人取得联系。本平台出于学术交流目的引用,无意侵犯原作者权益。如权利人认为不妥,请及时联系公众号后台,我们将立即删除或协商解决。
医学国自然,省自然,博士课题设计,医学实验外包,医学SCI,实验方案设计,免费的线上博导一对一沟通,确认实力后再谈合作,科研合作可以加微信:SCI971SCI
国家杰青一对一答疑视频
博士课题设计专家答疑,如何从亚专业中选取更适合的方向
医学国自然面上项目专家答疑实况,解决申请过程中的任何问题
医学省自然申请答疑,立项的关键条件是哪一些?从哪些方向可以杀出重围
国家科技重大专项,专家一对一指导方案设计实录
临床型博士如何准备国青标书?没有预实验怎么办?专家一对一解答规划
科研信息合集
高分文献解析
国自然立项
展博课题申报
医学硕博论文解析
肿瘤领域科研分享
麻醉领域科研分享
消化领域科研分享
骨科领域科研分享
中医药科研研究
100 项与 ROCK抑制剂 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 青光眼 | 药物发现 | 中国 | 2024-07-25 | |
| 肿瘤 | 药物发现 | 中国 | 2024-07-25 | |
| 骨质疏松症 | 药物发现 | 中国 | 2024-07-25 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
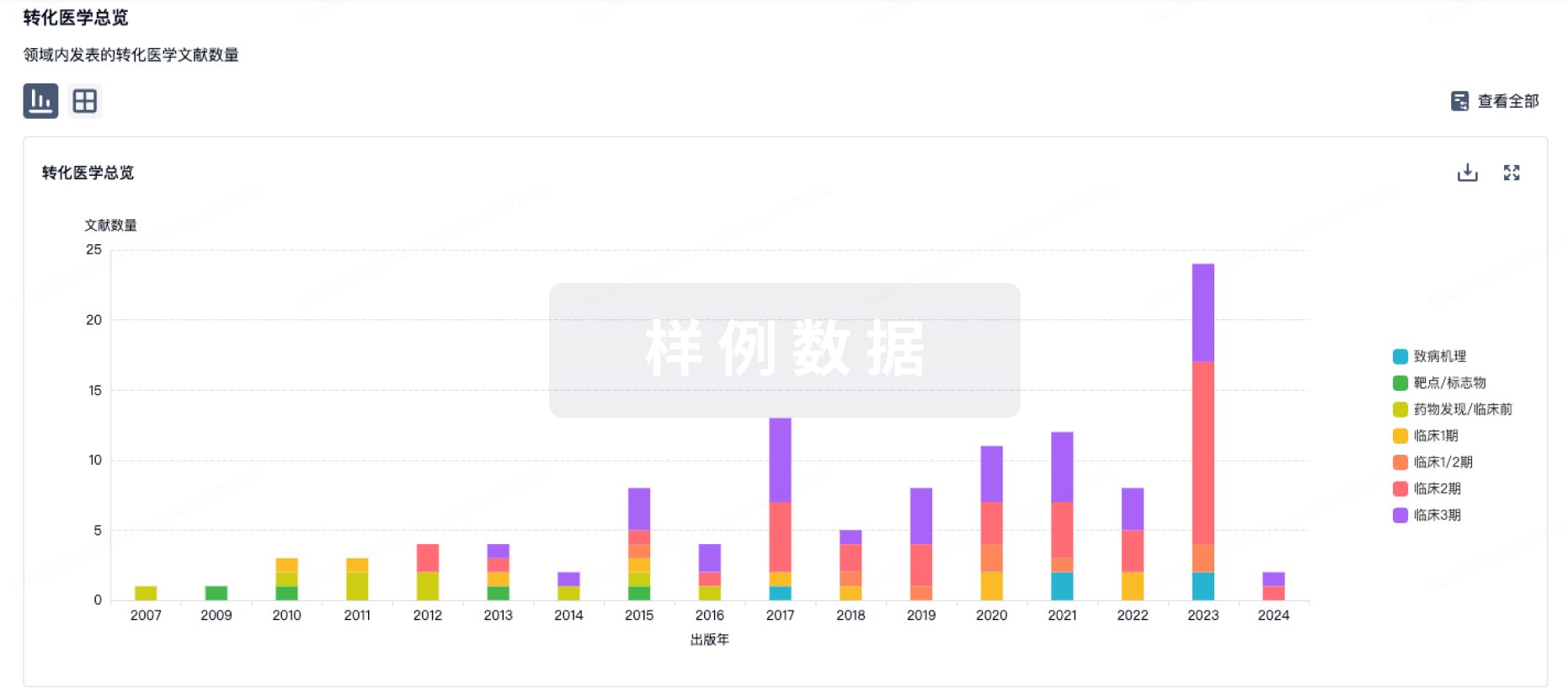
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
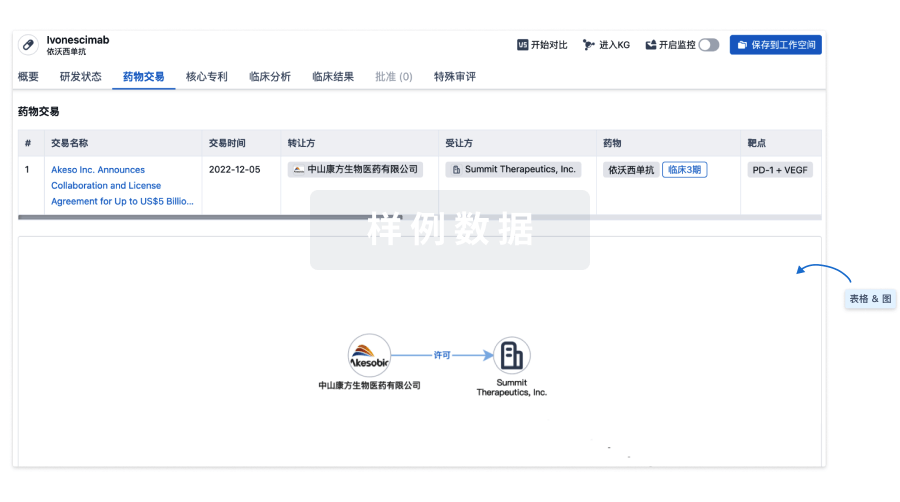
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
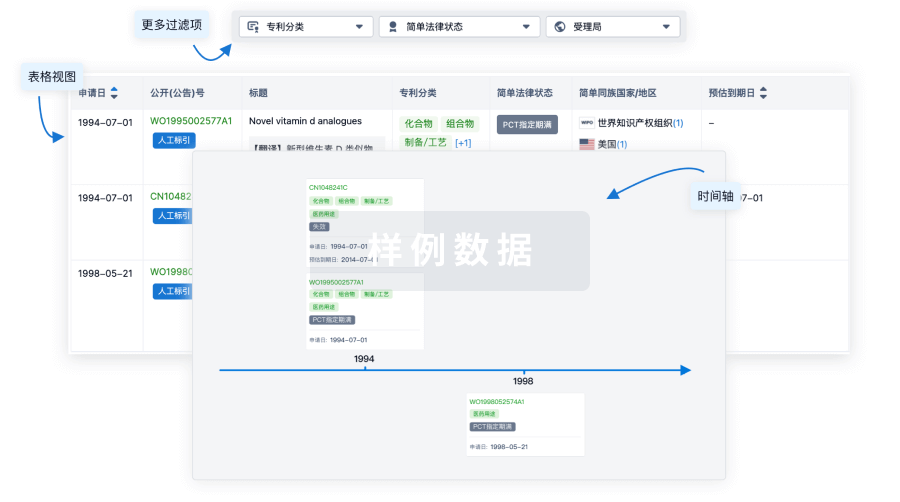
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
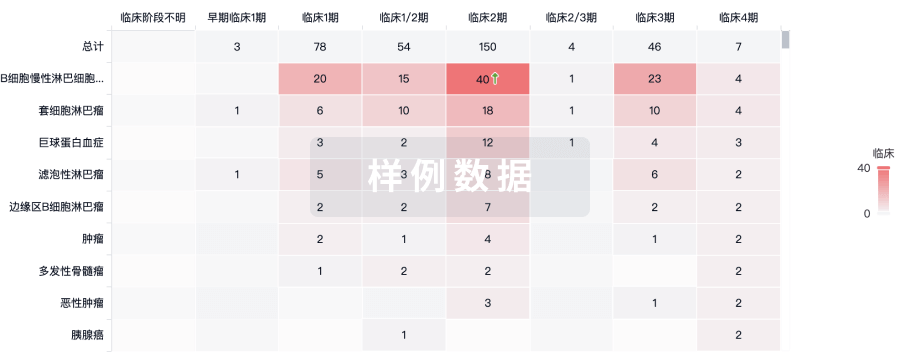
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用