预约演示
更新于:2026-02-07
GQ-ODN
更新于:2026-02-07
概要
基本信息
在研机构- |
权益机构- |
最高研发阶段无进展药物发现 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |

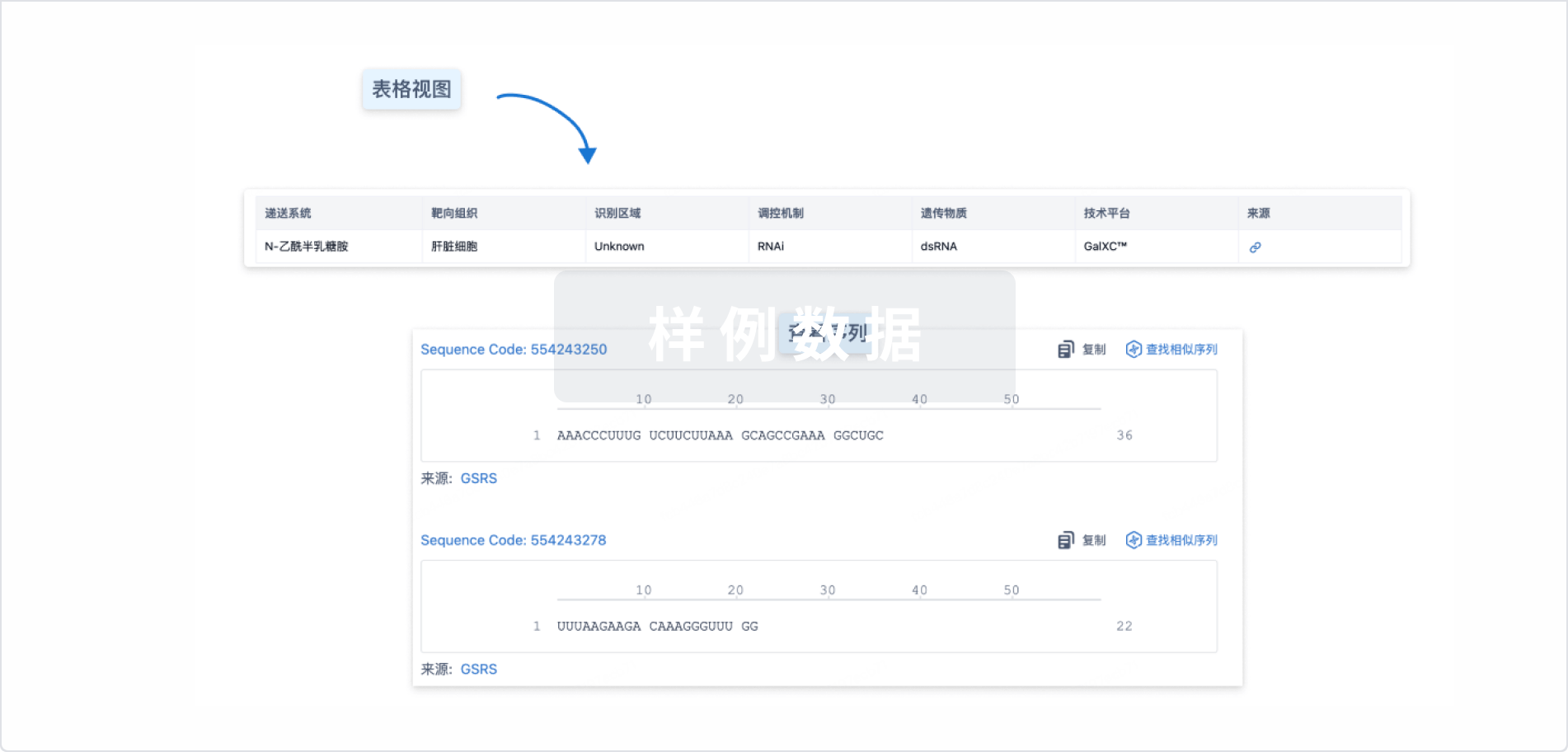
结构/序列
使用我们的RNA技术数据为新药研发加速。
登录
或
关联
100 项与 GQ-ODN 相关的临床结果
登录后查看更多信息
100 项与 GQ-ODN 相关的转化医学
登录后查看更多信息
100 项与 GQ-ODN 相关的专利(医药)
登录后查看更多信息
15
项与 GQ-ODN 相关的文献(医药)2014-05-01·Leukemia & lymphoma
Nanoparticle delivery of inhibitory signal transducer and activator of transcription 3 G-quartet oligonucleotides blocks tumor growth inHMGA1transgenic model of T-cell leukemia
Letter
作者: Naijie Jing ; David L. Huso ; Amy M. Belton ; James Turkson ; Francescopaolo Di Cello ; David J. Tweardy ; Linda M.S. Resar ; Sandeep N. Shah ; Joelle Hillion
While survival from childhood acute lymphoblastic leukemia (ALL) has improved significantly over the past three decades, pediatric patients with T-cell ALL (T-ALL) remain at high risk for relapse and poor outcomes compared to children with B-cell precursor ALL [1–3]. Outcomes for adults with T-ALL are even worse, with long-term survival rates of only 30–40% in patients less than 60 years, which falls to 10% in patients over 60 years [1–3]. Thus, studies are urgently needed to identify key molecular pathways that could be targeted in therapy. Similar to other malignancies, oncogene activation by mutation or overexpression is common in T-ALL. For example, overexpression of the TAL-1 oncogene occurs in most cases of pediatric T-ALL [2–4]. We discovered that the high mobility group A1 (HMGA1) oncogene is also overexpressed in T-ALL [1]. Activating mutations in other oncogenes have been identified, including LMO2, TLX1/HOX1, TLX3/HOX11L2 and HOXA, which appear to specify distinct T-ALL subtypes [1–3]. Other recurrent mutations in T-ALL include chromosomal loss of the CDKN2A/B tumor suppressor loci or activating mutations in the NOTCH1 pathway that occur in most subtypes of T-ALL [1–3]. NKX2-1 and MEF2C are putative oncogenes found in T-ALL subtypes that lack known oncogenic rearrangements [4]. Activating mutations in genes regulating cytokine receptor and RAS signaling were discovered in about two-thirds of all cases of an aggressive subclass of T-ALL, designated early T-cell precursor ALL [5]. This study also uncovered inactivating mutations in genes involved in normal hematopoietic development as well as histone modifying lesions. Intriguingly, the spectrum of lesions was similar to that observed in myeloid tumors and includes genes expressed in normal hematopoietic stem cells. An investigation of transcriptional networks in T-ALL identified RUNX1 as a tumor suppressor induced by TLX1 or TLX3 [6]. Together, these studies suggest that T-ALL is associated with diverse molecular underpinnings, which could contribute to the high rates of relapse with current therapies.
In addition to T-ALL, the HMGA1 oncogene is over-expressed in hematologic malignancies and solid tumors, including precursor B-ALL, refractory acute myeloid leukemia, and cancers of the breast, lung, colon, pancreas, uterus, brain, bladder and skin [1,7–12]. In fact, high expression of HMGA1 in leukemic blasts correlates with relapse in childhood B-ALL [7]. HMGA1 also induces leukemic transformation in cultured cells and causes aggressive leukemia in transgenic mice [1,7]. In a recent report, we found that HMGA1 cooperates with loss of function of the Cdkn2a (INK4A/ARF) tumor suppressor locus in a mouse model of T-ALL [1]. HMGA1 is also overexpressed in diverse, poorly differentiated solid tumors [7,12]. The HMGA1 gene encodes the HMGA1a and HMGA1b chromatin remodeling proteins, which function in modulating gene expression by altering chromatin structure [7]. HMGA1 proteins are the most abundant, non-histone, chromatin binding proteins found in cancer cells, and high expression portends a poor prognosis in diverse tumor types [7]. HMGA1 is also highly expressed during embryogenesis and in embryonic stem cells, hematopoietic stem cells, and cancer stem cells, including leukemic stem cells [7–12]. A recent landmark study revealed that HMGA1 is essential for reprogramming somatic cells into induced pluripotent stem cells by the four Yamanaka factors [10]. In addition, blocking HMGA1 expression or function interferes with multiple cancer phenotypes, including uncontrolled proliferation, anchorage-independent cell growth, migration, invasion, xenograft tumorigenesis and tumor progression in murine models [7,11–12]. HMGA1 is also required for cancer stem cell properties, such as three-dimensional sphere formation and limiting dilution tumorigenesis [11]. The transcriptional networks regulated by HMGA1 are emerging, and include genes involved in embryonic or adult stem cells, inflammation, signal transduction, cellular motility and hematopoiesis [7,10–12]. We previously reported that HMGA1 induces expression of the gene encoding the signal transducer and activator of transcription 3 (STAT3), and recent studies underscore a central role for STAT3 in inflammation, malignant transformation, tumor progression and a stem-like state [7,12–14]. STAT3 overexpression is also a prominent finding in hematologic malignancies [7,12]. Our prior studies demonstrate that blocking STAT3 function leads to apoptosis ex vivo in T-ALL cells from our HMGA1 transgenic model [12]. In addition, blocking HMGA1 or STAT3 function prevents colony formation in Burkitt leukemia cells [12]. Together, these studies suggest that targeting the HMGA1–STAT3 pathway could be an effective therapeutic approach in lymphoid tumors and other malignancies.
To determine whether blocking STAT3 function has anti-tumor activity in vivo in T-ALL induced by HMGA1, we treated HMGA1 transgenic mice that are deficient in the Cdkn2a tumor suppressor locus (HMGA1a–Cdkn2a−/−) with G-quartet oligodeoxynucleotides (GQ-ODNs) that specifically inhibit STAT3 binding to DNA [13,14]. GQ-ODNs were originally identified at the 3’ end of telomeres in vertebrates where they inhibit telomerase activity [13,14]. GQ-ODNs form intra- and inter-molecular four-stranded structures, or G-quartets, and block protein–DNA binding. GQ-ODNs ({"type":"entrez-protein","attrs":{"text":"T40214","term_id":"7491594","term_text":"pir||T40214"}}T40214) that inhibit STAT3 DNA binding were identified and found to have potent anti-tumor effects in experimental models of solid tumors, although they had not been tested in hematologic malignancies [13,14]. We therefore tested STAT3 GQ-ODN nanoparticles comprised of a 20 base-pair duplex DNA conjugated with polyethyleneimine (PEI) as previously described [13,14]. Prior to treatment, we confirmed that the STAT3 GQ-ODNs block STAT3 binding in gel shift experiments [Figure 1(A)]. As shown, the STAT3 GQ-ODN ({"type":"entrez-protein","attrs":{"text":"T40214","term_id":"7491594","term_text":"pir||T40214"}}T40214) prevents formation of the STAT3–DNA binding complex. In contrast, the control GQ-ODN had no effect on the STAT3–DNA complex. Next, we treated HMGA1a–Cdkn2a mice with the STAT3 GQ-ODN nanoparticles or control ODN nanoparticles. Because HMGA1a–Cdkn2a mice develop aggressive T-ALL by 20 weeks of age and succumb to their disease by 24–28 weeks, we began treatment at 20–21 weeks of age and sacrificed mice after 2 weeks of therapy (22–23 weeks of age) to assess tumor burden. Based on the GQ-ODN half-life of 48–72 h, mice were treated twice weekly with GQ-ODN nanoparticles delivered intraperitoneally following conjugation with PEI. To assess tumor burden following treatment, we compared relative splenic weights (spleen weight/total body weight) in mice treated with STAT3 GQ-ODN or control GQ-ODN nanoparticles, because previous studies from our group found that relative spleen weight is the best surrogate for tumor burden in these mice [1]. We found a marked decrease in tumor burden in the mice treated with STAT3 GQ-ODN [Figures 1(B) and 1(C)]. As shown, the spleens from STAT3 GQ-ODN-treated mice were smaller and lacked the grossly visible tumor nodules observed in the mice treated with control nanoparticles. Histopathologic analysis showed that the STAT3 GQ-ODN-treated mice had more normal splenic architecture and significantly smaller tumor burdens as compared to mice treated with control GQ-ODN [Figure 1(D)]. These studies indicate that blocking STAT3 with GQ-ODNs could be effective in hematologic malignancies with HMGA1 overexpression and high levels of activated STAT3, such as T-ALL. Our results further underscore the key role of STAT3 in leukemogenesis induced by HMGA1.
Figure 1
STAT3 inhibitory GQ-ODNs block STAT3 DNA-binding activity and exhibit anti-tumor properties in a murine model of aggressive T-cell leukemia. (A) STAT3 inhibitory GQ-ODNs block STAT3 DNA-binding activity in vSrc-transformed NIH3T3 cells. Gel shift experiments ...
In summary, we report for the first time that nanoparticle delivery of STAT3 GQ-ODNs has anti-leukemia effects in an HMGA1 transgenic model of aggressive T-ALL. Recent studies demonstrate a central role for HMGA1 in poorly differentiated or refractory cancers, embryonic stem cells and cellular reprogramming to an induced pluripotent stem cell [10,11]. Similarly, STAT3 signaling has been linked to inflammatory pathways, tumor progression and stem cell properties [7]. In fact, prior functional studies in cultured cell and murine models of solid tumors indicate that both HMGA1 and STAT3 are required for cancer stem cell properties, including growth as three-dimensional spheres and tumor-initiator or cancer stem cells [7]. Together, these data suggest that HMGA1 drives leukemic transformation and refractory disease by inducing STAT3 and stem-like transcriptional networks. Further studies will be needed to determine whether the HMGA1–STAT3 network is required for leukemic stem cell properties. In preliminary studies, we found enrichment of HMGA genes in leukemic stem cells from primary acute myeloid leukemia samples (Resar, unpublished data). In this brief report, we provide compelling evidence that blocking STAT3 function could be effective in T-cell lymphoid tumors driven, at least in part, by aberrant expression of HMGA1. Although further studies are needed, these findings indicate that the HMGA1–STAT3 pathway plays an important role in T-ALL in this model and could serve as a rational therapeutic target. Future studies are also needed to explore the role of STAT3 GQ-ODNs in other models of hematologic malignancies characterized by up-regulation or activation of HMGA1 and STAT3. Given the central role for HMGA1 as a master regulator in diverse cancers, targeting this oncogene directly should also have profound anti-tumor effects [11].
2014-02-01·Digestive diseases and sciences3区 · 医学
STAT3 and Importins Are Novel Mediators of Early Molecular and Cellular Responses in Experimental Duodenal Ulceration
3区 · 医学
Article
作者: Tetyana Khomenko ; Zsuzsanna Sandor ; Sandor Szabo ; Amrita Ahluwalia ; Xiaoming Deng ; Khushin N. Patel ; Andrzej Tarnawski
OBJECTIVES:
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that directly upregulates VEGF, Ref-1, p21, and anti-apoptotic genes such as Bcl-xL. In this study, we hypothesized that STAT3 signaling is activated and provides a critical protective role that is required for enterocyte survival during the early phases of cysteamine-induced duodenal ulcers.
METHODS:
We studied the effect of inhibition of STAT3 activity on cysteamine-induced duodenal ulcers in rats and egr-1 knockout mice using STAT3/DNA binding assay, immunohistochemistry, immunoblot, and quantitative reverse transcriptase PCR analyses.
RESULTS:
We found that G-quartet oligodeoxynucleotides T40214, a specific inhibitor of STAT3/DNA binding, aggravated cysteamine-induced duodenal ulcers in rats 2.8-fold (p < 0.05). In the pre-ulcerogenic stage, cysteamine induced STAT3 tyrosine phosphorylation, its translocation to nuclei, an increased expression and nuclear translocation of importin α and β in the rat duodenal mucosa. Cysteamine enhanced the binding of STAT3 to its DNA consensus sequences at 6, 12, and 24 h after cysteamine by 1.5-, 1.8-, and 3.5-fold, respectively, and activated the expression of STAT3 target genes such as VEGF, Bcl-xL, Ref-1, and STAT3-induced feedback inhibitor, a suppressor of cytokine signaling 3. We also demonstrated that egr-1 knockout mice, which are more susceptible to cysteamine-induced duodenal ulcers, had lower levels of STAT3 expression, its phosphorylation, expression of importin α or β, and STAT3/DNA binding than wild-type mice in response to cysteamine.
CONCLUSIONS:
Thus, STAT3 represents an important new molecular mechanism in experimental duodenal ulceration.
2013-12-01·Nucleic acids research2区 · 生物学
In-cell optical imaging of exogenous G-quadruplex DNA by fluorogenic ligands
2区 · 生物学
ArticleOA
作者: Tseng, Ting-Yuan ; Wang, Zi-Fu ; Chang, Ta-Chau ; Chien, Cheng-Hao
Abstract:
Guanine-rich oligonucleotides (GROs) are promising therapeutic candidate for cancer treatment and other biomedical application. We have introduced a G-quadruplex (G4) ligand, 3,6-bis(1-methyl-4-vinylpyridinium) carbazole diiodide, to monitor the cellular uptake of naked GROs and map their intracellular localizations in living cells by using confocal microscopy. The GROs that form parallel G4 structures, such as PU22, T40214 and AS1411, are detected mainly in the lysosome of CL1-0 lung cancer cells after incubation for 2 h. On the contrary, the GROs that form non-parallel G4 structures, such as human telomeres (HT23) and thrombin binding aptamer (TBA), are rarely detected in the lysosome, but found mainly in the mitochondria. Moreover, the fluorescence resonant energy transfer studies of fluorophore-labeled GROs show that the parallel G4 structures can be retained in CL1-0 cells, whereas the non-parallel G4 structures are likely distorted in CL1-0 cells after cellular uptake. Of interest is that the distorted G4 structure of HT23 from the non-parallel G4 structure can reform to a probable parallel G4 structure induced by a G4 ligand in CL1-0 living cells. These findings are valuable to the design and rationale behind the possible targeted drug delivery to specific cellular organelles using GROs.
100 项与 GQ-ODN 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 肿瘤 | 药物发现 | 美国 | - |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

特殊审评
只需点击几下即可了解关键药物信息。
登录
或

生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用