预约演示
更新于:2025-12-06
SMN-ASO
更新于:2025-12-06
概要
基本信息
非在研机构- |
权益机构- |
最高研发阶段临床前 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |
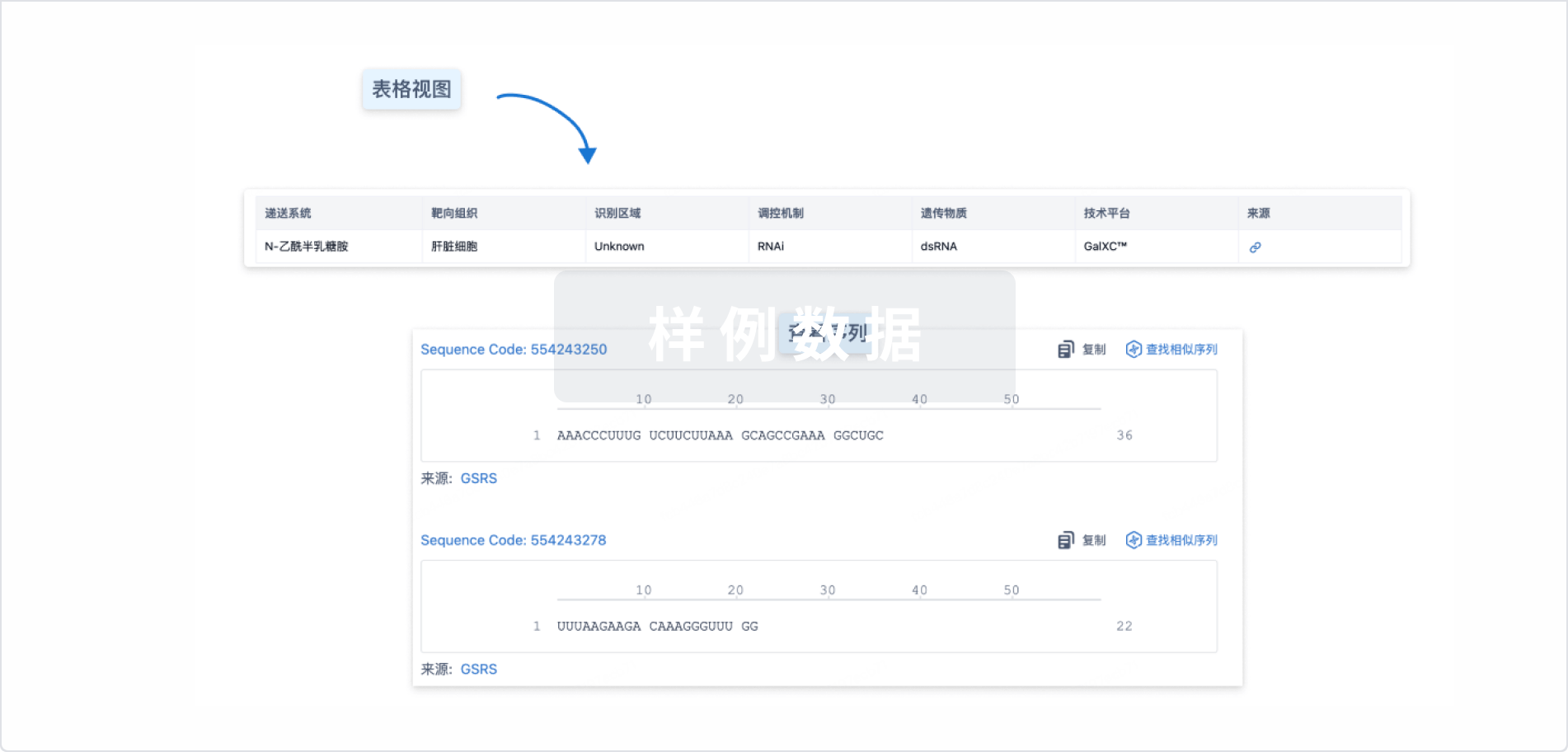
结构/序列
使用我们的RNA技术数据为新药研发加速。
登录
或
关联
100 项与 SMN-ASO 相关的临床结果
登录后查看更多信息
100 项与 SMN-ASO 相关的转化医学
登录后查看更多信息
100 项与 SMN-ASO 相关的专利(医药)
登录后查看更多信息
4
项与 SMN-ASO 相关的文献(医药)2022-09-01·Neurobiology of disease
Combinatorial ASO-mediated therapy with low dose SMN and the protective modifier Chp1 is not sufficient to ameliorate SMA pathology hallmarks
Article
作者: Janzen, E ; Rigo, F ; Rombo, R ; Ling, K K ; Muinos-Bühl, A ; Bennett, C F ; Wirth, B ; Hupperich, K
Spinal muscular atrophy (SMA) is a devastating genetically inherited neuromuscular disorder characterized by the progressive loss of motor neurons in the spinal cord, leading to muscle atrophy and weakness. Although SMA is caused by homozygous mutations in SMN1, the disease severity is mainly determined by the copy number of SMN2, an almost identical gene that produces ~10% correctly spliced SMN transcripts. Recently, three FDA- and EMA-approved therapies that either increase correctly spliced SMN2 transcripts (nusinersen and risdiplam) or replace SMN1 (onasemnogen abeparvovec-xioi) have revolutionized the clinical outcome in SMA patients. However, for severely affected SMA individuals carrying only two SMN2 copies even a presymptomatic therapy might be insufficient to fully counteract disease development. Therefore, SMN-independent compounds supporting SMN-dependent therapies represent a promising therapeutic approach. Recently, we have shown a significant amelioration of SMA disease hallmarks in a severely affected SMA mouse carrying a mutant Chp1 allele when combined with low-dose of SMN antisense oligonucleotide (ASO) treatment. CHP1 is a direct interacting partner of PLS3, a strong protective modifier of SMA. Both proteins ameliorate impaired endocytosis in SMA and significantly restore pathological hallmarks in mice. Here, we aimed to pharmacologically reduce CHP1 levels in an ASO-based combinatorial therapy targeting SMN and Chp1. Chp1 modulation is a major challenge since its genetic reduction to ~50% has shown to ameliorate SMA pathology, while the downregulation below that level causes cerebellar ataxia. Efficacy and tolerability studies determined that a single injection of 30 μg Chp1-ASO4 in the CNS is a safe dosage that significantly reduced CHP1 levels to ~50% at postnatal day (PND)14. Unfortunately, neither electrophysiological predictors such as compound muscle action potential (CMAP) or motor unit number estimation (MUNE) nor histological hallmarks of SMA in neuromuscular junction (NMJ), spinal cord or muscle were ameliorated in SMA mice treated with Chp1-ASO4 compared to CTRL-ASO at PND21. Surprisingly, CHP1 levels were almost at control level 4-weeks post injection, indicating a rather short-term effect of the ASO. Therefore, we re-administrated Chp1-ASO4 by i.c.v. bolus injection at PND28. However, no significant improvement of SMA hallmarks were seen at 2 month-of-age either. In conclusion, in contrast to the protective effect of genetically-induced Chp1 reduction on SMA, combinatorial therapy with Chp1- and SMN-ASOs failed to significantly ameliorate the SMA pathology. Chp1-ASOs compared to SMN-ASO proved to have rather short-term effect and even reinjection had no significant impact on SMA progression, suggesting that further optimization of the ASO may be required to fully explore the combination.
2020-12-01·Journal of pharmacological sciences3区 · 医学
Survival motor neuron protein regulates oxidative stress and inflammatory response in microglia of the spinal cord in spinal muscular atrophy
3区 · 医学
ArticleOA
作者: Shimazawa, Masamitsu ; Osanai, Daiki ; Nakamura, Shinsuke ; Takahashi, Kei ; Ando, Shiori ; Hara, Hideaki
The deficiency of survival motor neuron (SMN) protein can result in the onset of spinal muscular atrophy (SMA), an autosomal recessive disorder characterized by a progressive loss of motor neurons and skeletal muscle atrophy. The mechanism underlying SMA pathology remains unclear. Here, we demonstrate that SMN protein regulates oxidative stress and inflammatory response in microglia. Antisense oligonucleotide, which increases SMN protein expression (SMN-ASO), attenuated SMA model mice phenotypes and suppressed the activation of microglia in the spinal cord. The expression of oxidative stress marker in microglia was decreased by SMN-ASO injection in SMA model mice. Increased reactive oxygen species production and subsequent antioxidative stress reaction was observed in SMN protein-depleted RAW264.7. Furthermore, nuclear factor kappa B (NFκB) and c-Jun amino terminal kinase (JNK) signaling, which mainly mediate the inflammatory response, are activated in SMN protein-depleted RAW264.7. Tumor necrosis factor-α (TNF-α) production is also increased in SMN protein-depleted RAW264.7. These findings suggest that SMN protein regulates oxidative stress and inflammatory response in microglia, supporting current claims that microglia can be an effective target for SMA therapy.
2019-07-01·American journal of human genetics
NCALD Antisense Oligonucleotide Therapy in Addition to Nusinersen further Ameliorates Spinal Muscular Atrophy in Mice
Article
作者: Schneider, Svenja ; Bennett, C Frank ; Ling, Karen K ; Upadhyay, Aaradhita ; Wirth, Brunhilde ; Rigo, Frank ; Rombo, Roman ; Kononenko, Natalia L ; Torres-Benito, Laura ; Grysko, Vanessa
Spinal muscular atrophy (SMA) is a neuromuscular disease causing the most frequent genetic childhood lethality. Recently, nusinersen, an antisense oligonucleotide (ASO) that corrects SMN2 splicing and thereby increases full-length SMN protein, has been approved by the FDA and EMA for SMA therapy. However, the administration of nusinersen in severe and/or post-symptomatic SMA-affected individuals is insufficient to counteract the disease. Therefore, additional SMN-independent therapies are needed to support the function of motoneurons and neuromuscular junctions. We recently identified asymptomatic SMN1-deleted individuals who were protected against SMA by reduced expression of neurocalcin delta (NCALD). NCALD reduction is proven to be a protective modifier of SMA across species, including worm, zebrafish, and mice. Here, we identified Ncald-ASO3-out of 450 developed Ncald ASOs-as the most efficient and non-toxic ASO for the CNS, by applying a stepwise screening strategy in cortical neurons and adult and neonatal mice. In a randomized-blinded preclinical study, a single subcutaneous low-dose SMN-ASO and a single intracerebroventricular Ncald-ASO3 or control-ASO injection were presymptomatically administered in a severe SMA mouse model. NCALD reduction of >70% persisted for about 1 month. While low-dose SMN-ASO rescues multiorgan impairment, additional NCALD reduction significantly ameliorated SMA pathology including electrophysiological and histological properties of neuromuscular junctions and muscle at P21 and motoric deficits at 3 months. The present study shows the additional benefit of a combinatorial SMN-dependent and SMN-independent ASO-based therapy for SMA. This work illustrates how a modifying gene, identified in some asymptomatic individuals, helps to develop a therapy for all SMA-affected individuals.
100 项与 SMN-ASO 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 脊髓性肌萎缩 | 临床前 | 美国 | 2023-02-20 | |
| 脊髓性肌萎缩 | 临床前 | 德国 | 2023-02-20 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
No Data | |||||||
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

特殊审评
只需点击几下即可了解关键药物信息。
登录
或

生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用