预约演示
更新于:2026-05-12

Biofire Diagnostics LLC
更新于:2026-05-12
概览
关联
16
项与 Biofire Diagnostics LLC 相关的临床试验TCTR20250205003
Impact of implementing rapid diagnostic testing (multiplex PCR-based blood culture identification) among hospitalized pediatric patients at King Chulalongkorn Memorial Hospital
开始日期2024-10-06 |
申办/合作机构 |
NCT05314816
Clinical Impact of a Multiplex PCR Blood Culture Identification Panel in Early Identification of Positive Blood Cultures in Pediatric Patients in Guatemala
The purpose of this study is to assess the clinical impact of a rapid multiplex PCR blood culture identification panel on time to optimal antimicrobial therapy when compared to conventional microbiological culture methods in children hospitalized in a low resource setting in Guatemala City.
开始日期2022-04-07 |
申办/合作机构 |

NCT03933878
Rapid Detection of Airway Pathogens for Lung Transplantation
Pneumonias and lower respiratory tract infections can have important long-term consequences, particularly in the context of lung transplantation, where pneumonia is a major cause of death. Candidate organs and lung transplant recipients undergo bronchoscopic inspection to assess for lower respiratory tract infection, but traditional culture methods take time, leading to increased risk from inappropriate therapy. The investigators hypothesize that the rapid detection of lower respiratory tract infection, using a semi-quantitative multiplex molecular assay, can decrease the time to appropriate clinical decision making.
开始日期2019-03-19 |
申办/合作机构 |

100 项与 Biofire Diagnostics LLC 相关的临床结果
登录后查看更多信息
0 项与 Biofire Diagnostics LLC 相关的专利(医药)
登录后查看更多信息
15
项与 Biofire Diagnostics LLC 相关的文献(医药)2020-08-01·Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology
Clinical evaluation of the BioFire® Respiratory Panel 2.1 and detection of SARS-CoV-2
Article
作者: Jesse L. Cox ; Usha Spaulding ; Barbara Cabrera ; M.J. Broadhurst ; S.H. Hinrichs ; Margarita Rogatcheva ; Andy Schnaubelt ; Daria Drobysheva ; Allison M. Cushman-Vokoun ; Hannah M. Creager ; Salika M. Shakir ; Kevin M. Bourzac ; Alexander L. Greninger ; P.D. Fey ; Keith R. Jerome ; Keith D. Tardif ; Meei-Li Huang
We evaluated the performance of the BioFire® Respiratory Panel 2.1 (RP2.1) in the detection of SARS CoV-2 in comparison against three other SARS CoV-2 EUA assays. In these studies, the RP2.1 panel had 98 % positive percent agreement (48/49) and 100 % negative percent agreement (49/49). Since 30 % of nasopharyngeal swab specimens have a SARS CoV-2 Ct >30 and thus detection of virus in low titers is clinically relevant, a sample with a high titer was diluted and each 10 fold dilution was tested in triplicate and compared against 6 other EUA approved SARS CoV-2 assays. These data suggested that the BioFire® RP2.1 panel, along with four other SARS CoV-2 assays (Roche cobas, Cepheid Xpert Xpress, BioFire® Defense COVID19, and NECoV19), consistently detected viral RNA at the 10-7 dilution. Overall, these studies suggest that the BioFire® RP2.1 assay can be used to detect acute cases of SARS CoV2 in addition to patients with low viral titer later in disease presentation.
2014-03-01·The Journal of molecular diagnostics : JMD3区 · 医学
Getting Things Backwards to Prevent Primer Dimers
3区 · 医学
Article
作者: Ririe, Kirk M ; Poritz, Mark A
This Commentary highlights the article by Satterfield that describes a new class of primer technology-cooperative primers, which prevent primer-dimer amplification.
2013-11-18·Human antibodies
Developing and mature human granulocytes express ELP 6 in the cytoplasm
Article
作者: Wang, Shuping ; Christensen, Clarissa ; Spangrude, Gerald J. ; Perkins, Sherrie J. ; Wagner, Lori A. ; Dunn, Diane M. ; Wayner, Elizabeth A. ; Weiss, Robert B. ; Ward, Gavin W. ; Redd, Michael J. ; Gleich, Gerald J.
BACKGROUND:
c3orf75 is a conserved open reading frame within the human genome and has recently been identified as the Elongator subunit, ELP6 [1]. The Elongator enzyme complex has diverse roles, including translational control, neuronal development, cell migration and tumorigenicity [2].
OBJECTIVE:
To identify genes expressed early in human eosinophil development.
METHODS:
Eosinophilopoiesis was investigated by gene profiling of IL-5 stimulated CD34+ cells; ELP6 mRNA is upregulated. A monoclonal antibody was raised to the recombinant protein predicted by the open reading frame.
RESULTS:
ELP6 transcripts are upregulated in a human tissue culture model of eosinophil development during gene profiling experiments. Transcripts are expressed in most tissue types, as shown by reverse-transcriptase PCR. Western blot experiments show that human ELP6 is a 30 kDa protein expressed in the bone marrow, as well as in many other tissues. Flow cytometry experiments of human bone marrow mononuclear cells show that ELP6 is expressed intracellularly, in developing and mature human neutrophils, eosinophils and monocytes.
CONCLUSIONS:
ELP6 is expressed intracellularly in developing and mature granulocytes and monocytes but not in lymphocytes and erythrocytes.
32
项与 Biofire Diagnostics LLC 相关的新闻(医药)2026-02-25
真菌专题,第(9)篇。目录在文末。这是发表在 Clin Epidemiol. 2024 Aug 28:16:549-566.的一篇Review文章。摘要侵袭性念珠菌病(IC)是一种日益普遍、代价高昂且可能致命的感染,由机会性酵母菌念珠菌引起。此前,IC 主要由通常对药物敏感的白色念珠菌引起。全球范围内,由白色念珠菌引起的感染率呈下降趋势,而非白色念珠菌种的感染率上升,且耐药性增强,这给治疗带来了挑战。随着恶性肿瘤治疗手段的进步,IC 的高危人群数量增加,突破性 IC 感染的发病率也随之上升。此外,耳念珠菌的出现给管理和预防带来了诸多挑战,因其耐药性强且在医疗环境中传播迅速。尽管新型抗真菌药物的研发令人鼓舞,但了解 IC 流行病学的变化是未来管理和预防的关键一步。关键词:念珠菌、流行病学、耐药性、新出现的、非白色念珠菌属引言侵袭性念珠菌病(IC),在本综述中定义为念珠菌属在血液中(念珠菌血症)或从无菌组织中分离出念珠菌属(如肝脾念珠菌病和腹腔内念珠菌病),仍是一种代价高昂、致病性强且往往致命的感染。念珠菌血症已被发现会使 90 天死亡率增加超过 28%。1 为便于本综述,光滑假丝酵母(Nakaseomyces glabrata)将被称为光滑念珠菌(Candida glabrata),而克鲁斯假丝酵母(Pichia kudriavzevii)将被称为克鲁斯念珠菌(Candida krusei),因为这些名称在临床上仍被广泛使用。自 20 世纪 90 年代以来,已采取了多种策略以降低侵袭性感染的发生率,包括对高危人群进行抗真菌预防以及采取感染预防策略以降低医院内感染率。然而,尽管总体发病率有所下降,但新的挑战也随之出现,包括非白色念珠菌属念珠菌的增多、新型菌种如耳念珠菌(C. auris)的出现、突破性感染以及耐药性问题。2 本文将对侵袭性念珠菌病的流行病学变化及其对治疗的影响进行综述。念珠菌血症和侵袭性念珠菌病的临床表现念珠菌感染的临床表现多样,从黏膜局部感染到伴有败血症的严重播散性感染不等。念珠菌属被认为是人体皮肤、胃肠道和泌尿生殖道微生物群的正常组成部分。当宿主防御功能受损或微生物群失衡导致念珠菌过度生长时,就会发生感染(图 1)。图1. 侵袭性念珠菌病的常见危险因素。危险因素:静脉导管、全胃肠外营养、术后、广谱抗生素、非无菌部位定植、化疗、器官移植受者。宿主免疫反应黏膜持续暴露于念珠菌属微生物,因此进化出了高度协调的免疫反应,以实现宿主耐受并防止感染。上皮细胞是抵御感染的重要屏障,念珠菌通过诸如凝集素样序列(Als)蛋白家族等真菌黏附素附着于上皮细胞后,上皮细胞会检测到病原体相关分子模式(PAMPs),如甘露聚糖和1,3-β-D-葡聚糖。在上皮表面和全身性感染期间,免疫识别可能有所不同。Toll样受体和Dectin-1在侵袭性感染期间是宿主防御的重要且公认的组成部分,然而上皮细胞的反应可能需要非经典受体,如E-钙黏蛋白、EGFR/Her2和EphA1。这种反应必须同时杀灭真菌,同时尽量减少周围炎症反应并维持免疫稳态。这些宿主免疫的差异可能由念珠菌的存在形式所驱动,侵袭性感染中存在假菌丝形式,而黏膜表面则存在酵母形式。已鉴定出多种模式识别受体(PRR),包括 TLR2、TLR4、Dectin-1、FcγR、甘露糖受体、半乳糖凝集素 3、MINCLE 和 DC-SIGN。5 还观察到 CARD9 和 SYK 的下游信号传导在对念珠菌入侵的反应中至关重要。8 在这些基因组区域内的宿主多态性也已被鉴定,并且注意到这些多态性会增加宿主对感染的易感性。5组织驻留巨噬细胞在抗真菌防御中发挥关键作用,并产生炎性细胞因子和趋化因子以招募和激活包括中性粒细胞在内的其他免疫细胞。中性粒细胞的激活对于清除念珠菌至关重要,中性粒细胞减少是侵袭性疾病的主要危险因素。5 中性粒细胞还是唯一能够抑制念珠菌萌发的宿主细胞,感染的鼠类模型已明确表明中性粒细胞在念珠菌血症和/或侵袭性念珠菌病中的关键作用。9吞噬作用发生后,通过生成 NADPH 依赖性活性氧化物(ROS)来实现杀灭作用。该途径存在缺陷的患者(如慢性肉芽肿病患者)会出现侵袭性曲霉菌感染和其他病原体感染,但未观察到对念珠菌属的易感性增加。自然杀伤细胞在宿主防御念珠菌病方面似乎作用有限。树突状细胞是抵御真菌病原体的重要因素,对于处理和呈递真菌抗原以激活 T 细胞反应至关重要。T 细胞在宿主防御中也必不可少,CD4 和 CD8 细胞均能提供保护性免疫。Th 细胞产生的 Th-17 和 IFN-γ 可促进中性粒细胞和巨噬细胞的杀菌活性,该细胞反应通路中的定量缺陷(如 HIV)10 和定性差异(如宿主多态性)11 与各种形式的念珠菌病有关。临床表现局部黏膜皮肤感染包括鹅口疮(口腔念珠菌病)、食管和阴道酵母菌感染以及慢性黏膜皮肤念珠菌病。鹅口疮患者通常有基础糖尿病且血糖控制不佳、通过吸入方式局部使用糖皮质激素(如治疗哮喘)或为宿主反应不成熟的新生儿。食管感染可能与口腔鹅口疮同时发生,也可能单独出现,通常是更严重的潜在免疫缺陷的先兆,尤其是 T 细胞缺陷(如艾滋病、实体器官、造血干细胞移植受者、宿主反应不成熟的新生儿)。外阴阴道念珠菌病可能与上述任何基础疾病有关,也可能在近期使用抗生素后发生,因为抗生素会破坏保护性细菌种类。慢性黏膜皮肤感染见于特定基因组区域存在多态性的患者,特别是 STAT1 获得功能突变者12 或常染色体隐性遗传的多腺体自身免疫综合征 I 型患者13。相比之下,侵袭性疾病主要发生在宿主防御机制明显受损的人群中(图 1)。侵袭性感染的风险因素包括黏膜屏障严重受损(化疗后出现黏膜炎)且伴有中性粒细胞减少。这些患者还经常使用广谱抗生素,这会显著改变胃肠道微生物群,且常置有中心静脉导管——这些都是感染的额外风险因素。全胃肠外营养、血液透析、静脉注射毒品、胃肠道穿孔以及胃肠道手术均会增加侵袭性疾病的风险。14诊断目前可用诊断方法的敏感性和特异性极大地影响了我们对侵袭性念珠菌病(IC)流行病学的理解。IC 诊断的金标准是从血液或通常无菌的部位获得的阳性培养物。阳性结果可进行基质辅助激光解吸电离飞行时间(MALDI-TOF)质谱(MS)检测,以更快速地鉴定到种水平。15 然而,血液培养在后来经尸检证实患有 IC 的患者中仅呈阳性 21%至 71%。16 血液培养表现不佳可能与采集方法有关,特别是采集的血量,因为在念珠菌血症期间,每毫升血液中通常少于一个念珠菌菌落形成单位。17 但鉴于约三分之一的 IC 患者被归类为深部感染而无念珠菌血症,16 在所有 IC 病例中不能完全依赖血液培养。在这些患者中,诊断是通过受影响部位(如腹腔积液)的阳性培养物或组织病理学来确立的。综合来看,这些数据表明,血液培养对侵袭性念珠菌病的敏感性仅为 50%。16 血液培养效果相对较差,这促使人们开发了包括抗原检测18 和基于分子的方法在内的其他诊断手段。19已开发出几种检测念珠菌抗原(包括甘露聚糖和 1,3-β-D-葡聚糖)存在的检测方法。18 甘露聚糖抗原的诊断检测通常与抗甘露聚糖抗体检测同时进行,这种方法在儿科患者和中枢神经系统(CNS)感染患者中显示出有希望的结果,18 但在一项针对因严重腹部疾病而有念珠菌血症风险的非中性粒细胞减少性重症监护病房(ICU)患者的大型前瞻性研究中表现不佳。在该组患者中,甘露聚糖抗原的敏感性仅为 43.3%,特异性为 67.3%。20 抗甘露聚糖抗体检测的敏感性仅为 25.8%,但特异性更佳,为 89%。20 其他研究评估了联合检测甘露聚糖抗原和抗甘露聚糖抗体的性能,但敏感性仍不理想,仅为 51%,特异性为 71%。由于性能不佳,该检测未获美国食品药品监督管理局(FDA)批准。与甘露糖抗原检测相比,1,3-β-D-葡聚糖检测的性能特征有所改善,多项研究的平均灵敏度约为 85%,且阴性预测值通常高于 95%。18,22 然而,由于存在多种潜在的假阳性来源,包括:使用由纤维素制成的膜进行血液透析、接受静脉注射免疫球蛋白或白蛋白、同时使用抗菌药物、严重黏膜炎以及其他真菌感染,其特异性通常低于 60%。18已有多种基于分子的诊断方法获批使用,近期在其他文献中也有相关综述。19 与常用的 BioFire®FilmArray® 血培养鉴定(BCID)检测板(BioFire Diagnostics 公司,美国犹他州盐湖城)相比,T2Candida 检测板(T2 Biosystems 公司,美国马萨诸塞州列克星敦)无需血培养呈阳性。多项研究显示,T2Candida 检测板的敏感性为 91%,特异性为 94%。19 但该检测板仅限于检测五种念珠菌:白色念珠菌、热带念珠菌、克鲁斯念珠菌、光滑念珠菌和近平滑念珠菌。检测血浆中微生物游离 DNA(mcfDNA)的宏基因组下一代测序(mNGS)是一种很有前景的方法,有可能在其他血液生物标志物/检测仍为阴性时更早地检测和诊断真菌感染。23 到目前为止,通过这种方法诊断侵袭性念珠菌病的数据有限,尽管早期报告显示出潜力。24目前侵袭性念珠菌病的诊断方法显然存在局限性,这进而影响了我们对其流行病学的认识。不过令人鼓舞的是,相关研究仍在不断推进,改进和创新的技术有望实现更快速的诊断和治疗,同时也能提升流行病学评估的水平。侵袭性念珠菌病的负担在美国,念珠菌血症每年估计导致 22,000 例感染。25 念珠菌血症也是全国范围内导致医疗相关血流感染(BSIs)的第二大常见原因。26 老年人、男性以及黑人/非裔美国人中念珠菌血症的发病率较高。27,28 尽管念珠菌血症是侵袭性念珠菌病(IC)最常见的形式,但念珠菌属也可引起其他无菌或深部体位的感染,其中腹腔内念珠菌病(IAC)是重症监护病房(ICUs)中第二常见的 IC 类型。29–31 IAC 包含多种疾病表现,由于缺乏标准化的疾病定义,使得理解并准确把握这些感染的负担变得困难。29总体而言,美国侵袭性念珠菌病(IC)和念珠菌血症的发病率随时间推移呈下降趋势,并在近年来趋于平稳,这可能是因为感染控制措施的改进以及中心静脉导管护理包的实施。32 在美国,十年前 IC 的发病率较高,在 1996 至 2003 年期间,每 10 万人口中有 22 至 29 例感染。33 利用美国电子病历的最新数据显示,2009 至 2017 年间,IC 的发病率没有显著变化,住院患者 IC 的总体发病率为每 10 万例住院 90 例。34 利用来自主动人群监测的数据进行更细致的评估表明,2008 至 2017 年期间,美国多个地区的念珠菌血症发病率下降,25,27,35 同时,非血液来源(包括腹部无菌部位)的 IC 发病率从 2009 至 2017 年间有所上升。34尽管过去十年中念珠菌血症的总体发病率有所下降,但在全球新冠疫情背景下,其发病率却不幸上升。36–38 实际上,一些研究发现,感染新冠病毒的患者念珠菌血症的发病率高于未感染的患者。36–40 这些感染新冠病毒但无其他基础疾病的患者出现念珠菌血症,可能是由于与严重新冠病毒感染相关的医疗相关暴露所致。41 新冠疫情期间医疗系统的改变(例如,感染控制措施的疏漏、抗菌药物处方量的增加)以及感染新冠病毒的患者所需的高危护理(例如,侵入性设备、住院时间长),可能增加了新冠病毒感染患者发生侵袭性念珠菌病的风险。41,42尽管侵袭性念珠菌病(IC)在全球范围内仍是一种威胁,但其问题的严重程度难以评估。数据的可用性因实验室和监测能力以及分析方法的不同而有所差异。基于对 1990 年至 2016 年间 107 项欧洲研究数据的荟萃分析,念珠菌血症的总体合并发病率为每 10 万人中有 3.9 例。43 与美国的趋势相似,欧洲的研究报告称,2010 年后念珠菌血症的发病率有所下降,侵袭性念珠菌病(IAC)的发病率甚至更低,23 个欧洲重症监护病房(ICU)中 IAC 的发病率约为念珠菌血症发病率的三分之一。31,43 目前,亚洲、中东、非洲和拉丁美洲尚无基于人口的数据来源。44 然而,对中东和北非(MENA)地区有限数据的分析表明,卡塔尔的念珠菌血症发病率最高(每 10 万人中有 15.4 例),而伊朗的发病率最低(每 10 万人中有 0.3 例)。45 在亚洲,使用来自五个国家 25 家医院的实验室数据,念珠菌血症的发病率为每 1000 名患者中有 1.2 例。46,47 在南美洲,念珠菌血症的发病率在每 1000 次住院中为 0.6 至 6.0 例。48侵袭性念珠菌病(IC)与住院时间延长、医疗费用高昂以及发病率和死亡率上升有关。2019 年的一项研究显示,IC 导致美国 12770 例住院,平均每次住院损失 28 个工作日。50 据估计,IC 给美国造成的总经济负担为 18 亿美元。50 在美国,念珠菌血症的全因住院死亡率高达 36%。27,51,52 在欧洲,IC 的 30 天死亡率为 38%至 42%。31,43 亚洲地区的 IC 死亡率与之相当,据研究估计死亡率为 40%。46 在中东和北非地区,IC 死亡率的估计数据有限,但成人患者中死亡率在 33%至 60%之间。53 在南美洲,研究发现死亡率在 30%至 70%之间。48 在美国,IC 的全因死亡率在老年人中最高,在儿童中最低。25,27,52 没有报告种族或性别在死亡率方面存在显著差异。52IC 的总体负担很可能被低估了,尤其是考虑到全球范围内监测工作面临的挑战和存在的空白,以及现有诊断检测的性能特点。许多国家的诊断实验室检测和发现侵袭性念珠菌病(IC)的能力有限。缺乏标准化的方法和分母限制了对 IC 负担估计值进行比较的能力。44 此外,大多数 IC 研究都是单中心或规模较小的多中心分析。即使在拥有基于人口的监测系统的国家,念珠菌血症通常也不向公共卫生部门报告,因此报告是自愿的。尽管存在这些限制,但现有数据证实了 IC 给医疗保健带来的沉重负担以及与之相关的高患者死亡率。侵袭性念珠菌病的地域差异导致侵袭性念珠菌病(IC)或念珠菌血症的念珠菌属菌种的流行病学特征因地理区域而异。18 无论这些地域差异如何,全球范围内,白色念珠菌作为致病菌的比例明显呈下降趋势。在 20 世纪 80 年代至 90 年代,白色念珠菌的比例为 70%至 80%,54 而如今在大多数地理区域已降至 40%至 60%。25,55,56 尽管这种下降趋势普遍存在,但欧洲医学真菌学联合会(ECMM)开展的一系列多中心念珠菌研究显示,导致念珠菌血症的白色念珠菌比例从 1997 至 1999 年的 56.4%57 下降到 2006 至 2008 年的 54%,58 再到 2018 年的 46.2%,56 非白色念珠菌的比例相应上升(表 1),但存在重要的地域差异。在北欧和中欧,白色念珠菌的实际比例远高于 50%至 70%,而在南欧、拉丁美洲、澳大利亚和美国,其比例大多低于 50%。59,60表 1.欧洲医学真菌学联盟在三个不同时间点内导致念珠菌血症的致病菌种念珠菌物种1997–1999572006–200858201856C. albicans白色念珠菌56.4%54%46.0%C. glabrata光滑念珠菌13.9%13.8%21.0%C. parapsilosis假丝酵母菌13.5%18.5%13.0%C. tropicalis热带念珠菌7.2%6.0%7.0%C. krusei克柔假丝酵母菌2.0%2.5%3.0%其他7.0%5.2%10%非白色念珠菌属真菌分布的变化也存在明显的地区差异,在开具经验性治疗处方时,必须了解当地的流行病学模式,而不能仅仅依赖区域评估。18 光滑念珠菌的比例在美国、澳大利亚和北欧地区显著上升。25,40,41,55,61 相比之下,近平滑念珠菌的比例在亚洲(包括日本和中国)、拉丁美洲(包括巴西)和南欧地区有所增加。31,62–65 而热带念珠菌的比例在拉丁美洲和亚洲的大部分地区显著上升。62,66 总结整个欧洲的情况,光滑念珠菌的比例从 2006 至 2008 年的 13.8%58 上升到 2018 年的 21.4%56,这一变化十分显著。值得注意的是,念珠菌属真菌的分布不仅在不同地理区域之间存在差异,甚至在地理位置相近的医疗中心之间也可能有所不同,这或许受到包括既往抗真菌药物暴露和重症监护病房患者年龄在内的影响当地流行病学的因素的影响。67 除了已知病原体的全球分布外,念珠菌属新出现的菌种在全球范围内也存在差异。新型念珠菌物种随着全球气温的上升,真菌正在适应更高的环境温度,从而导致新的真菌物种作为人类病原体出现。最突出的例子是耳念珠菌,据推测它由植物腐生菌进化而来,可能在适应更高温度后成为人类病原体。这一假设因观察到环境分离株在哺乳动物体温下生长速度比临床菌株慢而得到加强,这一发现与它们的祖先最近适应了更高温度的观点一致。耳念珠菌如今已成为全球人类的威胁,尤其是在医疗保健环境中,在印度、南欧、英国、巴西和美国的重症监护病房引发了大规模疫情。因此,耳念珠菌被列为世界卫生组织最近发布的真菌病原体优先清单中的四种关键真菌病原体之一,这不仅是因为大规模疫情的数量,还因为该病原体具有多重耐药性。耳念珠菌的流行病学特征仍存在显著差异,一些国家仅报告零星病例,这可能是因为严格的医院卫生措施防止了院内传播和难以控制的暴发,75 而另一些国家则因这种多重耐药病原体引发的大规模暴发而处于紧急状态。75,76与耳念珠菌这种真正意义上的新兴病原体不同,之前被定义为“新兴”的念珠菌属物种(即念珠菌凯夫里亚种、念珠菌吉氏亚种、念珠菌卢西塔尼亚亚种、都柏林念珠菌、念珠菌法玛塔亚种、念珠菌隐匿亚种、念珠菌鲁戈萨亚种、念珠菌诺维吉尼亚亚种),实际上可能已经“出现”,这要么是因为诊断区分方法的改进,要么是因为广泛使用抗真菌治疗和预防措施导致的选择。例如,在引入基质辅助激光解吸电离飞行时间质谱(MALDI-TOF)后,都柏林念珠菌感染有所增加,因为该方法能够可靠地区分表型相似的白色念珠菌和都柏林念珠菌。其他一些菌株,如 C. inconspicua、C. norvegensis、C. guilliermondii、C. digboiensis 和 C. lusitaniae 对氟康唑和/或棘白菌素类药物具有固有的耐药性,因此可能仅在接触过抗真菌药物的患者群体中增多。尽管 C. parapsilosis 并非新兴菌种,但由耐氟康唑的 C. parapsilosis 引起的克隆性暴发在全球范围内呈上升趋势,令人担忧。携带 ERG11 Y132F 突变的耐氟康唑 C. parapsilosis 菌株可能不会遭受相关的适应性代价(甚至可能有生存优势),因此即使在没有三唑类药物暴露的情况下也可能大量繁殖。64 这种病原体与感染者的高死亡率相关,64 其具有在全球范围内广泛传播的潜在威胁。了解不仅新兴菌种,而且新兴耐药趋势的全球分布对于确定最佳的预防和治疗策略至关重要,尤其是在我们试图预防接受预防性抗真菌药物的易感人群发生突破性感染时。由于念珠菌导致的突破性感染由于念珠菌导致的突破性感染涵盖了一系列广泛的临床病症。突破性感染通常由对抗真菌药物具有耐药性的病原体引起,发生在具有多种风险因素、存在未被识别或未被清除的感染源以及/或者抗真菌药物药代动力学情况不佳的患者身上。直到最近,有关真菌感染的“突破性感染”这一术语在总体上仍由各个研究者自行解释,且定义模糊不清。最近,由真菌病研究组教育与研究联盟(MSGERC)和欧洲医学真菌学联合会(ECMM)共同发布的一份共识声明提出了突破性真菌感染(包括念珠菌病)的共识定义,这是朝着标准化此类观察迈出的重要一步。78 总体而言,突破性念珠菌感染在高危患者中并不常见,发生率低于 10%,79 尽管在某些特定病例系列中,此类感染可占所有念珠菌感染的 40%。80定义突破性(BT)念珠菌感染发生在个体接受预防性、经验性或抢先/靶向抗真菌治疗的情况下。与曲霉菌或地方性真菌等其他真菌感染不同,BT念珠菌感染的诊断需要从通常无菌的部位(如血液)获得阳性培养结果,或者从黏膜部位(如口咽部或食管)获得有说服力的培养和组织学证据。81 因此,根据欧洲癌症研究与治疗组织和真菌病研究组教育与研究联盟(EORTC/MSGERC)更新的标准,目前仅存在经证实的BT念珠菌感染。82 血清学证据,如念珠菌PCR、念珠菌甘露聚糖和抗甘露聚糖、1.3-β-D葡聚糖,可提供支持性证据,但不能作为念珠菌病的唯一证据来源。81 要符合BT的定义,感染必须发生在开始全身性抗真菌治疗至少72小时后,且在停用抗真菌药物后不超过一个给药间隔(8小时至7天,具体取决于所用抗真菌药物)。78突破性念珠菌病的风险因素与突破性念珠菌病相关的风险因素有若干种,其中许多事件发生在三个主要的宿主群体中:重症监护病房(ICU)长期住院患者、实体器官移植受者以及患有血液系统恶性肿瘤或接受干细胞移植的患者。79 这些宿主风险因素如下所述。宿主因素在三个主要的突破性感染风险群体中,存在一些共同的风险因素,包括存在中心静脉导管、长期(> 14 天)使用两种或两种以上广谱抗菌药物以及医源性免疫抑制。79 ICU 群体特有的风险因素包括 ICU 住院时间超过 10 天、并发胰腺炎、包括烧伤在内的重大创伤、肾功能受损、全胃肠外营养以及机械通气。83 尽管这些因素中的任何一种都可能增加这三个主要患者群体中任何一人的突破性感染风险,但在 ICU 人群中,它们的影响尤为显著,且可能具有协同作用。在移植人群中,肺、肝和小肠移植受者发生突破性念珠菌感染的风险增加是其特有的风险因素。80,84,85在迄今为止规模最大的研究中,TRANSNET 数据库确定,所有已确诊的念珠菌感染中有 41% 为血流感染型念珠菌病,且肺移植受者的感染率最高。在该系列研究中,抗真菌预防用药包括氟康唑、其他唑类药物、棘白菌素类药物和两性霉素 B。80 在患有血液系统恶性肿瘤和/或接受干细胞移植的患者中,血流感染型念珠菌病的特定危险因素包括黏膜炎、中性粒细胞减少、使用糖皮质激素和其他免疫抑制剂。79关于宿主因素的一个重要考虑是控制感染源。虽然这通常指的是对中心静脉导管或其他血管内装置进行适当的管理和移除,但这也可能涉及对腹腔脓肿、胸腔积脓或软组织脓肿等受污染的体液积聚物引流不畅的情况。79药代动力学因素血流感染型念珠菌病可能是药物浓度不足、药物在特定组织或空间内渗透不良或意外的药物相互作用所致。尽管遵循了推荐的给药方案,但唑类抗真菌药物的血药浓度不足最为常见。86因此,即使在进行抗真菌预防的情况下,也建议进行治疗药物监测,尤其是对伊曲康唑、伏立康唑和泊沙康唑的监测。87 同时,必须认识到一些常用药物对关键部位的渗透性有限。例如,棘白菌素类药物无法显著渗透到中枢神经系统或泌尿系统。最后,药物相互作用,尤其是三唑类药物与其他常联合使用的药物之间的相互作用,随着现代患者管理的复杂性不断增加而日益受到重视。诸如他克莫司、环孢素和西罗莫司等免疫抑制剂以及许多新型抗癌药物的种类繁多,这就需要详细了解这些可预测的药物相互作用,因为它们会显著影响抗真菌药物的疗效。88抗真菌耐药性对近期有关血流感染(BT)念珠菌感染的最新数据进行回顾表明,许多(但并非全部)此类感染是由对一种或多种抗真菌药物具有耐药性的病原体引起的,因此主要由非白色念珠菌属念珠菌引起,尤其是光滑念珠菌、克鲁斯念珠菌、近平滑念珠菌和热带念珠菌。89–92 较少见的菌种如路易斯安那念珠菌和吉氏念珠菌也有报道。93 大多数 BT 念珠菌感染发生在接受氟康唑、伏立康唑、泊沙康唑或艾沙康唑等三唑类抗真菌药物预防治疗的患者中,94–96 但也有大量报告称,一些患者在使用棘白菌素类药物时仍发生了突破性感染。97,98结局如果能早期识别并进行恰当处理,大多数 BT 念珠菌感染可通过抗真菌治疗成功治愈,包括优化剂量、控制感染源以及识别潜在的药物相互作用。在大多数大型系列研究中,BT 念珠菌感染与新发念珠菌感染在 30 天内的总体死亡率相似(约 30%)。99,100念珠菌属感染的流行趋势及其对抗真菌药物的敏感性变化至关重要,因为它们可能与非白色念珠菌属感染的流行病学变化以及抗真菌药物敏感性趋势有关。念珠菌属的流行趋势及抗真菌药物敏感性过去三十年对侵袭性念珠菌病(IC)的研究引发了对不断变化的抗真菌药物敏感性模式的大量调查,并记录了其随时间的变化。区域和全球范围内的调查记录了主要念珠菌属对抗真菌药物敏感性的趋势。27,101–104 在此讨论中,抗真菌药物敏感性是根据临床和实验室标准协会(CLSI)和/或欧洲抗菌药物敏感性试验委员会(EUCAST)的方法确定的,尽管这些方法和断点并非完全可互换,但其相似性足以进行一般性比较。105,106 接下来将分别对五种主要的念珠菌属以及耳念珠菌进行综述。在世界大多数地区,白色念珠菌仍是导致念珠菌血流感染及其他形式侵袭性念珠菌病的最常见原因。18,27,62,102,103,107–109 在过去二十年的多项人群研究中,白色念珠菌几乎普遍对氟康唑、棘白菌素类药物和两性霉素 B 敏感。对氟康唑耐药的情况很少见,耐药率通常在 0% 至 3% 之间,总体平均不到 1%。27,62,102,103,109在世界许多地区,光滑念珠菌是侵袭性念珠菌病的第二大常见致病菌,尤其影响老年人和有严重基础免疫抑制的患者。18 在常见的念珠菌属中,光滑念珠菌可能在抗真菌耐药性的发展方面构成最大风险,这可能归因于普遍存在的突变基因型,促进多药耐药表型。110 美国疾病控制与预防中心(CDC)开展的大规模人群调查显示,光滑念珠菌对棘白菌素类药物的耐药率在 2% 至 8% 之间,对氟康唑的耐药率在 7% 至 11% 之间。27在单中心调查中,光滑念珠菌对氟康唑的耐药率高达 75%,而在更大规模的研究中,对棘白菌素类药物的耐药率在 0% 至 24% 之间。27,111–113 亚历山大等人在一项为期 10 年的单中心调查中发现,棘白菌素耐药率与氟康唑耐药率的上升趋势大致相同。111 一般认为,学术机构中棘白菌素耐药率通常高于社区医疗中心,但并非所有中心都是如此。例如,最近一项来自一家大型学术医疗中心的调查在 10 年间检测了 800 多株光滑念珠菌,每年棘白菌素耐药菌株的比例均低于 3%,且未观察到耐药趋势。114 总体而言,过去 20 年来,光滑念珠菌对氟康唑和棘白菌素的耐药率缓慢但持续上升,对这一持续问题进行持续评估至关重要。与光滑念珠菌类似,热带念珠菌在世界不同地区的流行率差异很大。这种真菌在亚洲太平洋地区、加勒比地区和拉丁美洲尤为常见,但在北美和西欧则较少见。62 从历史上看,这种真菌对三唑类药物、棘白菌素类药物和两性霉素 B 均普遍敏感。然而,近年来对氟康唑和棘白菌素类药物耐药的报告日益增多,目前大多数地区对氟康唑的耐药基线水平为 3%至 5%,但在一些亚洲太平洋国家,这一比例高达 7%至 43%。115–117 大多数调查中对棘白菌素类药物的耐药率仍保持在 0%至 2%的较低水平。27,62 对两性霉素 B 耐药的情况仍然罕见,不过据印度的报告,泛耐药的热带假丝酵母菌可能占分离株的 1%。118在导致侵袭性念珠菌病的五种最常见的念珠菌属物种中,近平滑念珠菌的致死率(全因死亡率)最低。18 根据地理区域的不同,它是导致侵袭性念珠菌病的第一至第四常见原因。18,62,66 然而,在过去十年中,氟康唑耐药性已出现,某些系列中超过 50% 的近平滑念珠菌分离株耐药。64,119 在美国,氟康唑耐药率在 4% 至 10% 之间;而在世界其他地区,包括日本、西欧和拉丁美洲,这一比例要高得多。尽管由于 FKS1 基因的自然多态性,其对棘白菌素类药物的敏感性最低限值(MIC)比其他念珠菌属物种高,但对棘白菌素类药物的耐药性仍相对少见(0% 至 3%)。62,103,112,120克鲁斯念珠菌被认为对氟康唑固有耐药,但对伏立康唑和泊沙康唑的敏感性则有所不同。62,103,112 通常认为伏立康唑是治疗克鲁斯念珠菌感染的最佳口服药物选择,但最近的人群调查显示,伏立康唑的耐药率正在上升,在世界某些地区已接近 10%。棘白菌素和两性霉素 B 耐药性似乎也在出现,在较大的多中心研究中,耐药率在 3% 至 10% 之间。107,112自 2009 年在全球范围内出现以来,耳念珠菌在临床领域相对较新。虽然在美国是引起侵袭性念珠菌病的较少见原因,但该菌在印度和南非是念珠菌血流分离株中最常见的。118,121 它以潜在的多重抗真菌耐药性而著称,但数据显示,70% 至 90% 的分离株对氟康唑耐药,且对季铵盐消毒剂不敏感,这使得该病原体能够在医疗相关表面持续存在并引发暴发。122,123 对棘白菌素(高达 7%)和两性霉素 B(高达 35%)的耐药性在不同地理区域差异很大。122–124 在南亚的印度,对所有三类抗真菌药物耐药的情况最为常见(3%),而在世界其他地区则少见得多。122–124 由于耳念珠菌存在人与人之间的传播可能性,因此感染预防措施对于防止医院内传播至关重要。尽管许多念珠菌属菌种具有非常可预测的抗真菌药物敏感性模式(例如白色念珠菌),但对所有具有临床意义的分离株进行种水平鉴定并随后进行抗真菌药物敏感性检测仍然至关重要。125 如果常规检测不可用,应定期对足够数量的本地分离株进行表征,以便能够充分预测特定医院或地理区域中该种属的敏感性模式。IC 通用管理原则美国感染病学会(IDSA)125 及欧洲指南126,127 建议,在未鉴定至菌种水平之前,对于大多数侵袭性念珠菌病患者(无论是否中性粒细胞减少),首选棘白菌素类药物作为初始治疗,因其具有广谱活性、良好的副作用特征、降低死亡率的优势,以及非白色念珠菌属中唑类耐药率不断上升的情况。125,128–132 两份指南均建议,若临床症状改善且药敏试验或菌种鉴定未提示三唑类耐药,则在 5 - 7 天(IDSA)或 10 天(欧洲指南)后降级为氟康唑治疗。由于克鲁斯念珠菌对氟康唑固有耐药,因此对于该菌种,指南推荐伏立康唑作为降级治疗药物。125,133对于置有中心静脉导管的非中性粒细胞减少症患者,应拔除导管;但对于可能存在胃肠道念珠菌血症来源的中性粒细胞减少症患者,建议采取个体化的中心静脉导管拔除策略,但念珠菌近平滑念珠菌 IC.125 感染的情况除外。对于持续性念珠菌血症,建议进行超声心动图检查以评估是否存在心内膜炎。134 对于单纯性念珠菌血症,在血培养转阴且症状缓解后,治疗应持续 2 周。所有感染的心内装置均需移除或在疾病控制后进行无限期口服唑类药物抑制。125,132由于潜在的眼科并发症会对治疗产生重大影响,美国感染病学会(IDSA)建议,对于非中性粒细胞减少症患者,应在确诊后一周内进行散瞳眼底检查;对于中性粒细胞减少症患者,则应在中性粒细胞减少症缓解后一周内进行检查,因为直到免疫功能恢复,才可能出现脉络膜视网膜炎和眼内炎的证据。根据 IDSA 和欧洲指南,对于眼部受累情况,应联合眼科进行玻璃体内治疗,若对氟康唑或伏立康唑敏感,则使用这两种药物。否则,推荐使用玻璃体内两性霉素 B,可联合氟胞嘧啶,必要时考虑进行玻璃体切除术。治疗持续 4 至 6 周,或直至眼科复查结果恢复正常。125,132美国感染病学会(IDSA)建议,对于慢性播散性念珠菌病,应使用脂质体两性霉素 B 或棘白菌素类药物治疗数周,若对氟康唑敏感,则可逐步换用氟康唑,直至复查影像学显示病灶消退后方可停药。在高危化疗或干细胞移植期间,建议持续治疗以预防复发。对于骨髓炎,若出现大脓肿或关节不稳定,则需进行外科清创术。美国感染病学会(IDSA)建议使用口服唑类药物治疗 6 至 12 个月,或使用棘白菌素类药物(较弱的替代药物为脂质体两性霉素 B)治疗 2 周,随后改为口服唑类药物治疗 6 至 12 个月。欧洲指南倾向于使用口服唑类药物治疗 6 至 12 个月,或使用脂质体两性霉素 B 治疗 2 至 6 周,随后改为口服唑类药物完成 6 至 12 个月的疗程。对于化脓性关节炎,建议进行外科引流并移除任何人工装置,若装置保留,则需长期口服唑类药物进行抑制。IDSA 建议使用口服唑类药物治疗 6 周,或使用棘白菌素类药物治疗 2 周,随后使用口服唑类药物治疗≥4 周,或使用脂质体两性霉素 B 治疗 2 周,随后改为口服唑类药物治疗≥4 周。欧洲指南建议口服唑类药物治疗 6 周以上,或使用脂质体两性霉素 B 治疗 2 周,随后改为氟康唑治疗 4 周以上,伏立康唑治疗 6 周以上也可作为替代方案。125,132中枢神经系统(CNS)念珠菌病的最佳治疗方案数据较少。美国感染病学会(IDSA)建议使用脂质体两性霉素 B 联合或不联合氟胞嘧啶进行治疗,若临床反应良好且对氟康唑敏感,可考虑降阶梯治疗。治疗时长取决于症状、影像学及脑脊液异常的消退情况。正常剂量下棘白菌素类药物对中枢神经系统的渗透性较差,不推荐使用。125 欧洲指南弱推荐静脉注射脂质体两性霉素 B 联合氟胞嘧啶治疗 6 周,随后使用氟康唑治疗 3 周,或者使用脂质体两性霉素 B 联合氟康唑治疗 4 周,具体取决于药敏情况。132早期治疗的必要性侵袭性念珠菌病(IC)的归因死亡率仍然很高,且随着治疗的延迟而增加。念珠菌血培养平均需要 2 至 3 天才能生长,具体时间取决于菌种。因此,预防和经验性治疗策略均被常规采用以降低死亡率。133,135,136 IDSA 建议在侵袭性念珠菌病发生率超过 5%的重症监护病房(ICU)中,对高危患者考虑使用氟康唑或棘白菌素类药物进行预防。125,137一项针对非中性粒细胞减少性重症监护病房(ICU)患者预防措施的 Cochrane 综述未发现对死亡率有影响,但确实指出侵袭性真菌感染显著减少。138 欧洲指南建议对近期腹部手术后出现反复穿孔或渗漏的 ICU 患者使用氟康唑进行预防。132 对于异基因干细胞移植患者,在初始中性粒细胞减少期,推荐使用氟康唑、泊沙康唑、伏立康唑或米卡芬净进行预防,随后根据免疫恢复情况和移植物抗宿主病的免疫抑制情况,转为使用氟康唑或泊沙康唑。131美国感染病学会(IDSA)建议,对于有风险因素和感染替代标志物且病情恶化的患者,应考虑经验性抗真菌治疗。在出现感染性休克的情况下,应尽快开始治疗。不过,对于感染性休克且有侵袭性念珠菌病风险因素的患者,经验性抗真菌治疗的益处存在争议。EMPIRICUS 试验未能证明在有感染性休克和侵袭性念珠菌病风险因素的 ICU 患者中使用经验性米卡芬净的益处。139侵袭性念珠菌病的抗真菌药物研发管线尽管侵袭性念珠菌感染相关的发病率和死亡率仍然很高,但目前仅批准了四类主要的抗真菌药物用于全身治疗。棘白菌素类药物作为大多数念珠菌感染的一线治疗药物,在临床试验中侵袭性念珠菌病的总体失败率为 25% 至 30%,并且在泌尿道内几乎没有活性。18,133,140–144 由于多种机制(表 2),多重耐药念珠菌感染呈上升趋势,尤其是耳念珠菌,其对唑类药物广泛耐药,并且在治疗期间有可能对所有类别的抗真菌药物产生耐药性,这凸显了开发新药以应对流行病学变化的必要性。133,145–153表 2. 主要念珠菌属物种最常见的耐药机制念珠菌物种抵抗机制C. albicans白色念珠菌氟康唑——排出转运蛋白(MDR1、CDR1、CDR2),其氨基酸替代较少见C. glabrata光滑念珠菌阿唑类——药物排出体(Cdr1和Cdr2)的变化嵌棘白素——葡聚糖合酶基因中的FKS1多态性突变表型——不匹配修复缺陷导致多种抗真菌耐药性表型C. krusei克柔假丝酵母菌氟康唑——由于ERG11内部发生变化而具有内在耐药性,其他唑类常被保留C. parapsilosis假丝酵母菌氟康唑——ERG11中氨基酸取代(主要是Y132F),最常见的是对伏立康唑的敏感性不一C. lusitaniae葡萄牙假丝酵母两性霉素B——人工麦角甾醇合成基因(ERG3等)的突变或表达改变。C. auris(耳念珠菌)阿唑类——大多数为氟康唑耐药性(ERG11突变),且对其他唑类有不同易感性两性霉素B——假设由麦角甾醇生物合成的变化引起棘棘白素——由FKS1突变引起令人鼓舞的是,2023 年美国食品药品监督管理局批准了一种新的棘白菌素类药物——瑞扎芬净(rezafungin),用于治疗成人侵袭性真菌感染。在一项名为 ReSTORE 的 3 期临床试验中,瑞扎芬净与卡泊芬净(caspofungin)治疗侵袭性念珠菌病的疗效相当,治愈率达 60%。154,155 它的结构与阿尼芬净(anidulafungin)相似,但半衰期更长,可每周给药一次,这将使其更适合门诊治疗。149 目前,包括 ReSPECT 在内的其他研究正在进行中,ReSPECT 是一项针对接受异基因造血干细胞移植患者预防侵袭性真菌感染的 3 期临床试验。150,156伊布替芬净(Ibrexafungerp)是另一种新型药物,它在与棘白菌素不同的位点抑制(1→3)-β-D-葡聚糖合成,因此对许多因 FKS 突变而对棘白菌素耐药的菌株仍具有活性,包括许多耳念珠菌菌株。156 重要的是,它在酸性环境中仍具有溶解性和活性,使其适用于脓肿治疗。149,157 针对其他药物治疗无效的感染的临床试验结果令人鼓舞。150伊布替芬的口服生物利用度为 35% 至 50%,全身分布良好,但中枢神经系统除外。目前,伊布替芬仅获美国食品药品监督管理局批准用于治疗外阴阴道念珠菌病,不过针对侵袭性念珠菌病降阶梯治疗、难治性念珠菌病以及耳念珠菌的临床试验正在进行中。149,156–159最后,福沙匹坦(fosmanogepix)是另一种令人鼓舞的新型抗真菌药物。福沙匹坦是一种首创的 Gwt1 真菌蛋白抑制剂,Gwt1 蛋白类是将甘露糖蛋白锚定在细胞膜和细胞壁所必需的。160 这种新型机制对包括耐药念珠菌在内的多种念珠菌属具有广泛的活性。160 一项针对耳念珠菌所致念珠菌血症患者的 2 期临床试验结果表明,福沙匹坦安全有效。161 此外,一项针对由白色念珠菌、光滑念珠菌、近平滑念珠菌和/或都柏林念珠菌所致念珠菌血症患者的 2 期研究数据显示,20 名患者中有 16 名成功治愈,且未出现严重不良事件。162 3 期试验正在计划中,但鉴于抗真菌耐药率不断上升,这些初步结果令人鼓舞。医疗环境中的预防与感染控制鉴于与导管相关血流感染(CLABSI)相关的发病率和死亡率,预防是关键的优先事项。为此,美国感染病学会(IDSA)合作制定了多项干预措施的联合建议,以降低所有 CLABSI(包括念珠菌血症)的风险。163 这些 CLABSI 防护包的实施降低了感染的发生率。18 指南包括对置管适当指征的教育,指定特定人员并采用无菌技术来置管和护理中心静脉导管。上肢部位的感染风险低于下肢部位。163 所有置管部位应每日进行评估,如果出现静脉炎、感染或功能障碍的迹象,应移除外周导管。163 非隧道式锁骨下导管的感染风险低于颈内静脉或股静脉部位,建议使用最少数量的管腔。163 及时移除不必要的中心静脉导管可降低感染风险,所有置管和中心静脉导管的使用都应采用无菌技术和无菌屏障预防措施。163在置管前应进行氯己定皮肤清洁,根据敷料材质制定无菌敷料更换计划。只有在导管出现故障或怀疑感染时才更换中心静脉导管和中线导管,常规更换并不能降低感染率。163 对重症监护病房患者进行每日氯己定沐浴可降低包括念珠菌血症在内的血流感染风险。125,164医疗机构中耳念珠菌的暴发近期有所增加,这在很大程度上是由新冠疫情造成的,大量危重症患者需要机械通气,且感染控制措施可能出现了漏洞。145 该病原体在医疗机构环境中可长期存在,患者可能在一年以上仍处于定植状态。由于耳念珠菌在医疗机构中具有持续存在和传播的能力,美国疾病控制与预防中心发布了相关指南以限制其暴发。165,166 定植筛查是预防医疗机构中耳念珠菌传播的关键策略,通过识别未被察觉的定植者,从而采取适当的感染预防和控制措施。确定筛查对象取决于诸多因素,但应考虑对有高风险感染耳念珠菌的个体进行筛查,包括有流行病学关联的病例、在高风险机构有过接触史以及存在相关风险因素的患者。建议使用双侧腋窝和腹股沟的复合拭子进行筛查。感染耳念珠菌的患者应采取适当的隔离预防措施,这包括在急性护理医院采取接触预防措施,但在养老院环境中可考虑采取加强的屏障预防措施。由于患者通常会长期携带耳念珠菌,且检测结果可能时而呈阴性,因此在住院期间以及未来住院期间应持续采取隔离预防措施,不建议重复筛查以“清除”患者从而解除隔离预防措施。鉴于耳念珠菌在环境中的顽强生存能力,建议每天至少使用适当的消毒剂对感染患者的房间以及他们接触过的任何表面或共用设备(如血糖仪、移位机、体温计等)进行清洁。结论 侵袭性念珠菌病仍然是全球性的威胁。由于全球监测有限,再加上诊断方面的挑战,即便有新的临床诊断方法,该问题的严重程度可能仍被低估。尽管预防策略有所进步,但侵袭性念珠菌病仍是代价高昂且致病严重的疾病。虽然总体发病率似乎在下降,但治疗选择和感染控制措施对全球和局部的流行病学情况有着重大影响。非白色念珠菌属的增多以及多重耐药病原体 C. auris 的出现,将在不断演变的治疗实践中发挥重要作用,并影响该疾病的未来流行病学情况。尽管近期在治疗方面取得了进展,但鉴于突破性感染和耐药性的增加,仅靠抗真菌治疗在没有全面的念珠菌感染预防策略的情况下,不太可能成为有效的解决方案。需要在预防策略方面取得进展,并开发新的药物,以降低全球发病率并改善治疗效果。参考文献(略)。欢迎指正错误和交流。若光医学中心诺光门诊部(长沙)预约(下图)。中国女性洁净计划下图中二维码,可以扫码购买。、真菌专题(目录)(1)全球范围内抗真菌药物耐药性的出现对人类健康和粮食安全构成挑战(2)关于《微生物分子生物学评论》(MMBR)中有关真菌性行为的专题集:分子机制与进化意义(3)世界卫生组织真菌优先病原体清单成为游戏规则的改变者(4)针对世界卫生组织重点真菌病原体的新型治疗选择(5)耳念珠菌的遗传学与出现(6)全球严重真菌病的发病率和死亡率(7)在透析单位管理耳念珠菌:挑战、策略及未来方向(8)医疗机构中预防耳念珠菌传播的策略本文:(9)侵袭性念珠菌病的流行病学(10)耳念珠菌中心静脉导管相关血流感染在危重症患者中的情况:糟糕局面中的最坏情况(11)耳念珠菌感染(12)耳念珠菌:宿主相互作用、抗真菌药物耐药性及诊断(13)念珠菌病诊断与管理全球指南:欧洲临床微生物学与传染病学会(ECMM)与国际真菌病学会(ISHAM)及美国微生物学会(ASM)合作倡议(14)多重耐药念珠菌:流行病学、分子机制及治疗(15)抗真菌药物耐药性:白色念珠菌及其他物种中的分子机制(16)耳念珠菌:一种近期出现的多重耐药性真菌病原体(17)真菌细胞壁生物发生:结构复杂性、调控及抑制(18)单基因突变是新兴真菌病原体耳念珠菌代谢适应和获得丝状体能力的基础(19)Gcn5 赖氨酸乙酰转移酶介导耳念珠菌的细胞壁重塑、抗真菌药物耐药性以及毒力(20)抗 Hyr1p 单克隆抗体对多重耐药耳念珠菌所致系统性念珠菌病的保护效力(21)在“同一健康”的背景下理解抗真菌耐药性的临床和环境驱动因素(22)奥特塞康唑(Oteseconazole):复发性外阴阴道念珠菌病治疗的新进展(23)奥特塞康唑与氟康唑治疗严重外阴阴道念珠菌病的疗效比较:一项多中心、随机、双盲、Ⅲ期临床试验(24)奥特塞康唑:期待已久的抗真菌药物新成员,为复发性外阴阴道念珠菌病(RVVC)的治疗带来新选择(25)氟胞嘧啶及其临床应用(26)耳念珠菌会经历依赖黏附素和不依赖黏附素的细胞聚集(27)耳念珠菌中由 ACE2 缺失介导的细胞聚集调节皮肤真菌定植和宿主免疫反应(28)新出现的真菌病原体耳念珠菌诱导干扰素γ定植于哺乳动物毛囊(29)在“同一健康”的背景下理解抗真菌耐药性的临床和环境驱动因素(30)瑞扎芬净(瑞扎约)用于成人念珠菌血症和侵袭性念珠菌病治疗的审批中的监管考量(31)在治疗过程中体内出现高水平耐药性揭示了耳念珠菌对两性霉素 B 耐药性的首个已知机制(32)耳念珠菌细胞表面黏附素的功能冗余对于细胞间相互作用和聚集至关重要(33)新出现的真菌病原体耳念珠菌诱导IFNγ 以定植皮肤(34)干扰素-γ在自身免疫性多内分泌腺综合征 1 型中的作用(35)针对耳念珠菌这种真菌病原体的先天免疫反应(36)调控耳念珠菌病理生物学特征及抗真菌药物耐药性的信号通路(37)人体真菌病原体的免疫反应及治疗前景(38)针对耳念珠菌这种真菌病原体的先天免疫反应(39)耳念珠菌对氟胞嘧啶的体外快速耐药性进化(40)全基因组分析实验进化而来的耳念珠菌揭示了多种新型多重耐药机制(41)黏附素 Als4112 通过与角质形成细胞和细胞外基质蛋白的相互作用促进耳念珠菌皮肤定植(42)全球范围内抗真菌药物耐药性的出现对人类健康和粮食安全构成挑战(43)白色-棕色转换控制耳念珠菌的表型可塑性和毒力(44)抗真菌药物耐药性的分子机制(45)美国真菌病的经济负担~~等。
微生物疗法
2026-02-25
·若光医学
真菌专题,第(9)篇。目录在文末。
这是发表在 Clin Epidemiol. 2024 Aug 28:16:549-566.的一篇Review文章。
摘要
侵袭性念珠菌病(IC)是一种日益普遍、代价高昂且可能致命的感染,由机会性酵母菌念珠菌引起。此前,IC 主要由通常对药物敏感的白色念珠菌引起。全球范围内,由白色念珠菌引起的感染率呈下降趋势,而非白色念珠菌种的感染率上升,且耐药性增强,这给治疗带来了挑战。随着恶性肿瘤治疗手段的进步,IC 的高危人群数量增加,突破性 IC 感染的发病率也随之上升。此外,耳念珠菌的出现给管理和预防带来了诸多挑战,因其耐药性强且在医疗环境中传播迅速。尽管新型抗真菌药物的研发令人鼓舞,但了解 IC 流行病学的变化是未来管理和预防的关键一步。
关键词:念珠菌、流行病学、耐药性、新出现的、非白色念珠菌属
引言
侵袭性念珠菌病(IC),在本综述中定义为念珠菌属在血液中(念珠菌血症)或从无菌组织中分离出念珠菌属(如肝脾念珠菌病和腹腔内念珠菌病),仍是一种代价高昂、致病性强且往往致命的感染。念珠菌血症已被发现会使 90 天死亡率增加超过 28%。1 为便于本综述,光滑假丝酵母(Nakaseomyces glabrata)将被称为光滑念珠菌(Candida glabrata),而克鲁斯假丝酵母(Pichia kudriavzevii)将被称为克鲁斯念珠菌(Candida krusei),因为这些名称在临床上仍被广泛使用。自 20 世纪 90 年代以来,已采取了多种策略以降低侵袭性感染的发生率,包括对高危人群进行抗真菌预防以及采取感染预防策略以降低医院内感染率。然而,尽管总体发病率有所下降,但新的挑战也随之出现,包括非白色念珠菌属念珠菌的增多、新型菌种如耳念珠菌(C. auris)的出现、突破性感染以及耐药性问题。2 本文将对侵袭性念珠菌病的流行病学变化及其对治疗的影响进行综述。
念珠菌血症和侵袭性念珠菌病的临床表现念珠菌感染的临床表现多样,从黏膜局部感染到伴有败血症的严重播散性感染不等。念珠菌属被认为是人体皮肤、胃肠道和泌尿生殖道微生物群的正常组成部分。当宿主防御功能受损或微生物群失衡导致念珠菌过度生长时,就会发生感染(图 1)。
图1. 侵袭性念珠菌病的常见危险因素。危险因素:静脉导管、全胃肠外营养、术后、广谱抗生素、非无菌部位定植、化疗、器官移植受者。
宿主免疫反应
黏膜持续暴露于念珠菌属微生物,因此进化出了高度协调的免疫反应,以实现宿主耐受并防止感染。上皮细胞是抵御感染的重要屏障,念珠菌通过诸如凝集素样序列(Als)蛋白家族等真菌黏附素附着于上皮细胞后,上皮细胞会检测到病原体相关分子模式(PAMPs),如甘露聚糖和1,3-β-D-葡聚糖。在上皮表面和全身性感染期间,免疫识别可能有所不同。Toll样受体和Dectin-1在侵袭性感染期间是宿主防御的重要且公认的组成部分,然而上皮细胞的反应可能需要非经典受体,如E-钙黏蛋白、EGFR/Her2和EphA1。这种反应必须同时杀灭真菌,同时尽量减少周围炎症反应并维持免疫稳态。这些宿主免疫的差异可能由念珠菌的存在形式所驱动,侵袭性感染中存在假菌丝形式,而黏膜表面则存在酵母形式。
已鉴定出多种模式识别受体(PRR),包括 TLR2、TLR4、Dectin-1、FcγR、甘露糖受体、半乳糖凝集素 3、MINCLE 和 DC-SIGN。5 还观察到 CARD9 和 SYK 的下游信号传导在对念珠菌入侵的反应中至关重要。8 在这些基因组区域内的宿主多态性也已被鉴定,并且注意到这些多态性会增加宿主对感染的易感性。5
组织驻留巨噬细胞在抗真菌防御中发挥关键作用,并产生炎性细胞因子和趋化因子以招募和激活包括中性粒细胞在内的其他免疫细胞。中性粒细胞的激活对于清除念珠菌至关重要,中性粒细胞减少是侵袭性疾病的主要危险因素。5 中性粒细胞还是唯一能够抑制念珠菌萌发的宿主细胞,感染的鼠类模型已明确表明中性粒细胞在念珠菌血症和/或侵袭性念珠菌病中的关键作用。9
吞噬作用发生后,通过生成 NADPH 依赖性活性氧化物(ROS)来实现杀灭作用。该途径存在缺陷的患者(如慢性肉芽肿病患者)会出现侵袭性曲霉菌感染和其他病原体感染,但未观察到对念珠菌属的易感性增加。
自然杀伤细胞在宿主防御念珠菌病方面似乎作用有限。树突状细胞是抵御真菌病原体的重要因素,对于处理和呈递真菌抗原以激活 T 细胞反应至关重要。T 细胞在宿主防御中也必不可少,CD4 和 CD8 细胞均能提供保护性免疫。Th 细胞产生的 Th-17 和 IFN-γ 可促进中性粒细胞和巨噬细胞的杀菌活性,该细胞反应通路中的定量缺陷(如 HIV)10 和定性差异(如宿主多态性)11 与各种形式的念珠菌病有关。
临床表现
局部黏膜皮肤感染包括鹅口疮(口腔念珠菌病)、食管和阴道酵母菌感染以及慢性黏膜皮肤念珠菌病。鹅口疮患者通常有基础糖尿病且血糖控制不佳、通过吸入方式局部使用糖皮质激素(如治疗哮喘)或为宿主反应不成熟的新生儿。食管感染可能与口腔鹅口疮同时发生,也可能单独出现,通常是更严重的潜在免疫缺陷的先兆,尤其是 T 细胞缺陷(如艾滋病、实体器官、造血干细胞移植受者、宿主反应不成熟的新生儿)。外阴阴道念珠菌病可能与上述任何基础疾病有关,也可能在近期使用抗生素后发生,因为抗生素会破坏保护性细菌种类。慢性黏膜皮肤感染见于特定基因组区域存在多态性的患者,特别是 STAT1 获得功能突变者12 或常染色体隐性遗传的多腺体自身免疫综合征 I 型患者13。
相比之下,侵袭性疾病主要发生在宿主防御机制明显受损的人群中(图 1)。侵袭性感染的风险因素包括黏膜屏障严重受损(化疗后出现黏膜炎)且伴有中性粒细胞减少。这些患者还经常使用广谱抗生素,这会显著改变胃肠道微生物群,且常置有中心静脉导管——这些都是感染的额外风险因素。全胃肠外营养、血液透析、静脉注射毒品、胃肠道穿孔以及胃肠道手术均会增加侵袭性疾病的风险。14
诊断
目前可用诊断方法的敏感性和特异性极大地影响了我们对侵袭性念珠菌病(IC)流行病学的理解。IC 诊断的金标准是从血液或通常无菌的部位获得的阳性培养物。阳性结果可进行基质辅助激光解吸电离飞行时间(MALDI-TOF)质谱(MS)检测,以更快速地鉴定到种水平。15 然而,血液培养在后来经尸检证实患有 IC 的患者中仅呈阳性 21%至 71%。16 血液培养表现不佳可能与采集方法有关,特别是采集的血量,因为在念珠菌血症期间,每毫升血液中通常少于一个念珠菌菌落形成单位。17 但鉴于约三分之一的 IC 患者被归类为深部感染而无念珠菌血症,16 在所有 IC 病例中不能完全依赖血液培养。在这些患者中,诊断是通过受影响部位(如腹腔积液)的阳性培养物或组织病理学来确立的。综合来看,这些数据表明,血液培养对侵袭性念珠菌病的敏感性仅为 50%。16 血液培养效果相对较差,这促使人们开发了包括抗原检测18 和基于分子的方法在内的其他诊断手段。19
已开发出几种检测念珠菌抗原(包括甘露聚糖和 1,3-β-D-葡聚糖)存在的检测方法。18 甘露聚糖抗原的诊断检测通常与抗甘露聚糖抗体检测同时进行,这种方法在儿科患者和中枢神经系统(CNS)感染患者中显示出有希望的结果,18 但在一项针对因严重腹部疾病而有念珠菌血症风险的非中性粒细胞减少性重症监护病房(ICU)患者的大型前瞻性研究中表现不佳。在该组患者中,甘露聚糖抗原的敏感性仅为 43.3%,特异性为 67.3%。20 抗甘露聚糖抗体检测的敏感性仅为 25.8%,但特异性更佳,为 89%。20 其他研究评估了联合检测甘露聚糖抗原和抗甘露聚糖抗体的性能,但敏感性仍不理想,仅为 51%,特异性为 71%。由于性能不佳,该检测未获美国食品药品监督管理局(FDA)批准。
与甘露糖抗原检测相比,1,3-β-D-葡聚糖检测的性能特征有所改善,多项研究的平均灵敏度约为 85%,且阴性预测值通常高于 95%。18,22 然而,由于存在多种潜在的假阳性来源,包括:使用由纤维素制成的膜进行血液透析、接受静脉注射免疫球蛋白或白蛋白、同时使用抗菌药物、严重黏膜炎以及其他真菌感染,其特异性通常低于 60%。18
已有多种基于分子的诊断方法获批使用,近期在其他文献中也有相关综述。19 与常用的 BioFire®FilmArray® 血培养鉴定(BCID)检测板(BioFire Diagnostics 公司,美国犹他州盐湖城)相比,T2Candida 检测板(T2 Biosystems 公司,美国马萨诸塞州列克星敦)无需血培养呈阳性。多项研究显示,T2Candida 检测板的敏感性为 91%,特异性为 94%。19 但该检测板仅限于检测五种念珠菌:白色念珠菌、热带念珠菌、克鲁斯念珠菌、光滑念珠菌和近平滑念珠菌。检测血浆中微生物游离 DNA(mcfDNA)的宏基因组下一代测序(mNGS)是一种很有前景的方法,有可能在其他血液生物标志物/检测仍为阴性时更早地检测和诊断真菌感染。23 到目前为止,通过这种方法诊断侵袭性念珠菌病的数据有限,尽管早期报告显示出潜力。24
目前侵袭性念珠菌病的诊断方法显然存在局限性,这进而影响了我们对其流行病学的认识。不过令人鼓舞的是,相关研究仍在不断推进,改进和创新的技术有望实现更快速的诊断和治疗,同时也能提升流行病学评估的水平。
侵袭性念珠菌病的负担
在美国,念珠菌血症每年估计导致 22,000 例感染。25 念珠菌血症也是全国范围内导致医疗相关血流感染(BSIs)的第二大常见原因。26 老年人、男性以及黑人/非裔美国人中念珠菌血症的发病率较高。27,28 尽管念珠菌血症是侵袭性念珠菌病(IC)最常见的形式,但念珠菌属也可引起其他无菌或深部体位的感染,其中腹腔内念珠菌病(IAC)是重症监护病房(ICUs)中第二常见的 IC 类型。29–31 IAC 包含多种疾病表现,由于缺乏标准化的疾病定义,使得理解并准确把握这些感染的负担变得困难。29
总体而言,美国侵袭性念珠菌病(IC)和念珠菌血症的发病率随时间推移呈下降趋势,并在近年来趋于平稳,这可能是因为感染控制措施的改进以及中心静脉导管护理包的实施。32 在美国,十年前 IC 的发病率较高,在 1996 至 2003 年期间,每 10 万人口中有 22 至 29 例感染。33 利用美国电子病历的最新数据显示,2009 至 2017 年间,IC 的发病率没有显著变化,住院患者 IC 的总体发病率为每 10 万例住院 90 例。34 利用来自主动人群监测的数据进行更细致的评估表明,2008 至 2017 年期间,美国多个地区的念珠菌血症发病率下降,25,27,35 同时,非血液来源(包括腹部无菌部位)的 IC 发病率从 2009 至 2017 年间有所上升。34
尽管过去十年中念珠菌血症的总体发病率有所下降,但在全球新冠疫情背景下,其发病率却不幸上升。36–38 实际上,一些研究发现,感染新冠病毒的患者念珠菌血症的发病率高于未感染的患者。36–40 这些感染新冠病毒但无其他基础疾病的患者出现念珠菌血症,可能是由于与严重新冠病毒感染相关的医疗相关暴露所致。41 新冠疫情期间医疗系统的改变(例如,感染控制措施的疏漏、抗菌药物处方量的增加)以及感染新冠病毒的患者所需的高危护理(例如,侵入性设备、住院时间长),可能增加了新冠病毒感染患者发生侵袭性念珠菌病的风险。41,42
尽管侵袭性念珠菌病(IC)在全球范围内仍是一种威胁,但其问题的严重程度难以评估。数据的可用性因实验室和监测能力以及分析方法的不同而有所差异。基于对 1990 年至 2016 年间 107 项欧洲研究数据的荟萃分析,念珠菌血症的总体合并发病率为每 10 万人中有 3.9 例。43 与美国的趋势相似,欧洲的研究报告称,2010 年后念珠菌血症的发病率有所下降,侵袭性念珠菌病(IAC)的发病率甚至更低,23 个欧洲重症监护病房(ICU)中 IAC 的发病率约为念珠菌血症发病率的三分之一。31,43 目前,亚洲、中东、非洲和拉丁美洲尚无基于人口的数据来源。44 然而,对中东和北非(MENA)地区有限数据的分析表明,卡塔尔的念珠菌血症发病率最高(每 10 万人中有 15.4 例),而伊朗的发病率最低(每 10 万人中有 0.3 例)。45 在亚洲,使用来自五个国家 25 家医院的实验室数据,念珠菌血症的发病率为每 1000 名患者中有 1.2 例。46,47 在南美洲,念珠菌血症的发病率在每 1000 次住院中为 0.6 至 6.0 例。48
侵袭性念珠菌病(IC)与住院时间延长、医疗费用高昂以及发病率和死亡率上升有关。2019 年的一项研究显示,IC 导致美国 12770 例住院,平均每次住院损失 28 个工作日。50 据估计,IC 给美国造成的总经济负担为 18 亿美元。50 在美国,念珠菌血症的全因住院死亡率高达 36%。27,51,52 在欧洲,IC 的 30 天死亡率为 38%至 42%。31,43 亚洲地区的 IC 死亡率与之相当,据研究估计死亡率为 40%。46 在中东和北非地区,IC 死亡率的估计数据有限,但成人患者中死亡率在 33%至 60%之间。53 在南美洲,研究发现死亡率在 30%至 70%之间。48 在美国,IC 的全因死亡率在老年人中最高,在儿童中最低。25,27,52 没有报告种族或性别在死亡率方面存在显著差异。52
IC 的总体负担很可能被低估了,尤其是考虑到全球范围内监测工作面临的挑战和存在的空白,以及现有诊断检测的性能特点。许多国家的诊断实验室检测和发现侵袭性念珠菌病(IC)的能力有限。缺乏标准化的方法和分母限制了对 IC 负担估计值进行比较的能力。44 此外,大多数 IC 研究都是单中心或规模较小的多中心分析。即使在拥有基于人口的监测系统的国家,念珠菌血症通常也不向公共卫生部门报告,因此报告是自愿的。尽管存在这些限制,但现有数据证实了 IC 给医疗保健带来的沉重负担以及与之相关的高患者死亡率。
侵袭性念珠菌病的地域差异
导致侵袭性念珠菌病(IC)或念珠菌血症的念珠菌属菌种的流行病学特征因地理区域而异。18 无论这些地域差异如何,全球范围内,白色念珠菌作为致病菌的比例明显呈下降趋势。在 20 世纪 80 年代至 90 年代,白色念珠菌的比例为 70%至 80%,54 而如今在大多数地理区域已降至 40%至 60%。25,55,56 尽管这种下降趋势普遍存在,但欧洲医学真菌学联合会(ECMM)开展的一系列多中心念珠菌研究显示,导致念珠菌血症的白色念珠菌比例从 1997 至 1999 年的 56.4%57 下降到 2006 至 2008 年的 54%,58 再到 2018 年的 46.2%,56 非白色念珠菌的比例相应上升(表 1),但存在重要的地域差异。在北欧和中欧,白色念珠菌的实际比例远高于 50%至 70%,而在南欧、拉丁美洲、澳大利亚和美国,其比例大多低于 50%。59,60
表 1.欧洲医学真菌学联盟在三个不同时间点内导致念珠菌血症的致病菌种念珠菌物种1997–1999572006–200858201856C. albicans
白色念珠菌
56.4%
54%
46.0%C. glabrata光滑念珠菌
13.9%
13.8%
21.0%C. parapsilosis假丝酵母菌
13.5%
18.5%
13.0%C. tropicalis热带念珠菌
7.2%
6.0%
7.0%C. krusei克柔假丝酵母菌
2.0%
2.5%
3.0%
其他
7.0%
5.2%
10%
非白色念珠菌属真菌分布的变化也存在明显的地区差异,在开具经验性治疗处方时,必须了解当地的流行病学模式,而不能仅仅依赖区域评估。18 光滑念珠菌的比例在美国、澳大利亚和北欧地区显著上升。25,40,41,55,61 相比之下,近平滑念珠菌的比例在亚洲(包括日本和中国)、拉丁美洲(包括巴西)和南欧地区有所增加。31,62–65 而热带念珠菌的比例在拉丁美洲和亚洲的大部分地区显著上升。62,66 总结整个欧洲的情况,光滑念珠菌的比例从 2006 至 2008 年的 13.8%58 上升到 2018 年的 21.4%56,这一变化十分显著。值得注意的是,念珠菌属真菌的分布不仅在不同地理区域之间存在差异,甚至在地理位置相近的医疗中心之间也可能有所不同,这或许受到包括既往抗真菌药物暴露和重症监护病房患者年龄在内的影响当地流行病学的因素的影响。67 除了已知病原体的全球分布外,念珠菌属新出现的菌种在全球范围内也存在差异。
新型念珠菌物种
随着全球气温的上升,真菌正在适应更高的环境温度,从而导致新的真菌物种作为人类病原体出现。最突出的例子是耳念珠菌,据推测它由植物腐生菌进化而来,可能在适应更高温度后成为人类病原体。这一假设因观察到环境分离株在哺乳动物体温下生长速度比临床菌株慢而得到加强,这一发现与它们的祖先最近适应了更高温度的观点一致。耳念珠菌如今已成为全球人类的威胁,尤其是在医疗保健环境中,在印度、南欧、英国、巴西和美国的重症监护病房引发了大规模疫情。因此,耳念珠菌被列为世界卫生组织最近发布的真菌病原体优先清单中的四种关键真菌病原体之一,这不仅是因为大规模疫情的数量,还因为该病原体具有多重耐药性。耳念珠菌的流行病学特征仍存在显著差异,一些国家仅报告零星病例,这可能是因为严格的医院卫生措施防止了院内传播和难以控制的暴发,75 而另一些国家则因这种多重耐药病原体引发的大规模暴发而处于紧急状态。75,76
与耳念珠菌这种真正意义上的新兴病原体不同,之前被定义为“新兴”的念珠菌属物种(即念珠菌凯夫里亚种、念珠菌吉氏亚种、念珠菌卢西塔尼亚亚种、都柏林念珠菌、念珠菌法玛塔亚种、念珠菌隐匿亚种、念珠菌鲁戈萨亚种、念珠菌诺维吉尼亚亚种),实际上可能已经“出现”,这要么是因为诊断区分方法的改进,要么是因为广泛使用抗真菌治疗和预防措施导致的选择。例如,在引入基质辅助激光解吸电离飞行时间质谱(MALDI-TOF)后,都柏林念珠菌感染有所增加,因为该方法能够可靠地区分表型相似的白色念珠菌和都柏林念珠菌。其他一些菌株,如 C. inconspicua、C. norvegensis、C. guilliermondii、C. digboiensis 和 C. lusitaniae 对氟康唑和/或棘白菌素类药物具有固有的耐药性,因此可能仅在接触过抗真菌药物的患者群体中增多。
尽管 C. parapsilosis 并非新兴菌种,但由耐氟康唑的 C. parapsilosis 引起的克隆性暴发在全球范围内呈上升趋势,令人担忧。携带 ERG11 Y132F 突变的耐氟康唑 C. parapsilosis 菌株可能不会遭受相关的适应性代价(甚至可能有生存优势),因此即使在没有三唑类药物暴露的情况下也可能大量繁殖。64 这种病原体与感染者的高死亡率相关,64 其具有在全球范围内广泛传播的潜在威胁。了解不仅新兴菌种,而且新兴耐药趋势的全球分布对于确定最佳的预防和治疗策略至关重要,尤其是在我们试图预防接受预防性抗真菌药物的易感人群发生突破性感染时。
由于念珠菌导致的突破性感染
由于念珠菌导致的突破性感染涵盖了一系列广泛的临床病症。突破性感染通常由对抗真菌药物具有耐药性的病原体引起,发生在具有多种风险因素、存在未被识别或未被清除的感染源以及/或者抗真菌药物药代动力学情况不佳的患者身上。直到最近,有关真菌感染的“突破性感染”这一术语在总体上仍由各个研究者自行解释,且定义模糊不清。最近,由真菌病研究组教育与研究联盟(MSGERC)和欧洲医学真菌学联合会(ECMM)共同发布的一份共识声明提出了突破性真菌感染(包括念珠菌病)的共识定义,这是朝着标准化此类观察迈出的重要一步。78 总体而言,突破性念珠菌感染在高危患者中并不常见,发生率低于 10%,79 尽管在某些特定病例系列中,此类感染可占所有念珠菌感染的 40%。80
定义
突破性(BT)念珠菌感染发生在个体接受预防性、经验性或抢先/靶向抗真菌治疗的情况下。与曲霉菌或地方性真菌等其他真菌感染不同,BT念珠菌感染的诊断需要从通常无菌的部位(如血液)获得阳性培养结果,或者从黏膜部位(如口咽部或食管)获得有说服力的培养和组织学证据。81 因此,根据欧洲癌症研究与治疗组织和真菌病研究组教育与研究联盟(EORTC/MSGERC)更新的标准,目前仅存在经证实的BT念珠菌感染。82 血清学证据,如念珠菌PCR、念珠菌甘露聚糖和抗甘露聚糖、1.3-β-D葡聚糖,可提供支持性证据,但不能作为念珠菌病的唯一证据来源。81 要符合BT的定义,感染必须发生在开始全身性抗真菌治疗至少72小时后,且在停用抗真菌药物后不超过一个给药间隔(8小时至7天,具体取决于所用抗真菌药物)。78
突破性念珠菌病的风险因素
与突破性念珠菌病相关的风险因素有若干种,其中许多事件发生在三个主要的宿主群体中:重症监护病房(ICU)长期住院患者、实体器官移植受者以及患有血液系统恶性肿瘤或接受干细胞移植的患者。79 这些宿主风险因素如下所述。
宿主因素
在三个主要的突破性感染风险群体中,存在一些共同的风险因素,包括存在中心静脉导管、长期(> 14 天)使用两种或两种以上广谱抗菌药物以及医源性免疫抑制。79 ICU 群体特有的风险因素包括 ICU 住院时间超过 10 天、并发胰腺炎、包括烧伤在内的重大创伤、肾功能受损、全胃肠外营养以及机械通气。83 尽管这些因素中的任何一种都可能增加这三个主要患者群体中任何一人的突破性感染风险,但在 ICU 人群中,它们的影响尤为显著,且可能具有协同作用。
在移植人群中,肺、肝和小肠移植受者发生突破性念珠菌感染的风险增加是其特有的风险因素。80,84,85在迄今为止规模最大的研究中,TRANSNET 数据库确定,所有已确诊的念珠菌感染中有 41% 为血流感染型念珠菌病,且肺移植受者的感染率最高。在该系列研究中,抗真菌预防用药包括氟康唑、其他唑类药物、棘白菌素类药物和两性霉素 B。80 在患有血液系统恶性肿瘤和/或接受干细胞移植的患者中,血流感染型念珠菌病的特定危险因素包括黏膜炎、中性粒细胞减少、使用糖皮质激素和其他免疫抑制剂。79
关于宿主因素的一个重要考虑是控制感染源。虽然这通常指的是对中心静脉导管或其他血管内装置进行适当的管理和移除,但这也可能涉及对腹腔脓肿、胸腔积脓或软组织脓肿等受污染的体液积聚物引流不畅的情况。79
药代动力学因素
血流感染型念珠菌病可能是药物浓度不足、药物在特定组织或空间内渗透不良或意外的药物相互作用所致。尽管遵循了推荐的给药方案,但唑类抗真菌药物的血药浓度不足最为常见。86因此,即使在进行抗真菌预防的情况下,也建议进行治疗药物监测,尤其是对伊曲康唑、伏立康唑和泊沙康唑的监测。87 同时,必须认识到一些常用药物对关键部位的渗透性有限。例如,棘白菌素类药物无法显著渗透到中枢神经系统或泌尿系统。最后,药物相互作用,尤其是三唑类药物与其他常联合使用的药物之间的相互作用,随着现代患者管理的复杂性不断增加而日益受到重视。诸如他克莫司、环孢素和西罗莫司等免疫抑制剂以及许多新型抗癌药物的种类繁多,这就需要详细了解这些可预测的药物相互作用,因为它们会显著影响抗真菌药物的疗效。88
抗真菌耐药性
对近期有关血流感染(BT)念珠菌感染的最新数据进行回顾表明,许多(但并非全部)此类感染是由对一种或多种抗真菌药物具有耐药性的病原体引起的,因此主要由非白色念珠菌属念珠菌引起,尤其是光滑念珠菌、克鲁斯念珠菌、近平滑念珠菌和热带念珠菌。89–92 较少见的菌种如路易斯安那念珠菌和吉氏念珠菌也有报道。93 大多数 BT 念珠菌感染发生在接受氟康唑、伏立康唑、泊沙康唑或艾沙康唑等三唑类抗真菌药物预防治疗的患者中,94–96 但也有大量报告称,一些患者在使用棘白菌素类药物时仍发生了突破性感染。97,98
结局
如果能早期识别并进行恰当处理,大多数 BT 念珠菌感染可通过抗真菌治疗成功治愈,包括优化剂量、控制感染源以及识别潜在的药物相互作用。在大多数大型系列研究中,BT 念珠菌感染与新发念珠菌感染在 30 天内的总体死亡率相似(约 30%)。99,100念珠菌属感染的流行趋势及其对抗真菌药物的敏感性变化至关重要,因为它们可能与非白色念珠菌属感染的流行病学变化以及抗真菌药物敏感性趋势有关。
念珠菌属的流行趋势及抗真菌药物敏感性
过去三十年对侵袭性念珠菌病(IC)的研究引发了对不断变化的抗真菌药物敏感性模式的大量调查,并记录了其随时间的变化。区域和全球范围内的调查记录了主要念珠菌属对抗真菌药物敏感性的趋势。27,101–104 在此讨论中,抗真菌药物敏感性是根据临床和实验室标准协会(CLSI)和/或欧洲抗菌药物敏感性试验委员会(EUCAST)的方法确定的,尽管这些方法和断点并非完全可互换,但其相似性足以进行一般性比较。105,106 接下来将分别对五种主要的念珠菌属以及耳念珠菌进行综述。
在世界大多数地区,白色念珠菌仍是导致念珠菌血流感染及其他形式侵袭性念珠菌病的最常见原因。18,27,62,102,103,107–109 在过去二十年的多项人群研究中,白色念珠菌几乎普遍对氟康唑、棘白菌素类药物和两性霉素 B 敏感。对氟康唑耐药的情况很少见,耐药率通常在 0% 至 3% 之间,总体平均不到 1%。27,62,102,103,109
在世界许多地区,光滑念珠菌是侵袭性念珠菌病的第二大常见致病菌,尤其影响老年人和有严重基础免疫抑制的患者。18 在常见的念珠菌属中,光滑念珠菌可能在抗真菌耐药性的发展方面构成最大风险,这可能归因于普遍存在的突变基因型,促进多药耐药表型。110 美国疾病控制与预防中心(CDC)开展的大规模人群调查显示,光滑念珠菌对棘白菌素类药物的耐药率在 2% 至 8% 之间,对氟康唑的耐药率在 7% 至 11% 之间。27在单中心调查中,光滑念珠菌对氟康唑的耐药率高达 75%,而在更大规模的研究中,对棘白菌素类药物的耐药率在 0% 至 24% 之间。27,111–113 亚历山大等人在一项为期 10 年的单中心调查中发现,棘白菌素耐药率与氟康唑耐药率的上升趋势大致相同。111 一般认为,学术机构中棘白菌素耐药率通常高于社区医疗中心,但并非所有中心都是如此。例如,最近一项来自一家大型学术医疗中心的调查在 10 年间检测了 800 多株光滑念珠菌,每年棘白菌素耐药菌株的比例均低于 3%,且未观察到耐药趋势。114 总体而言,过去 20 年来,光滑念珠菌对氟康唑和棘白菌素的耐药率缓慢但持续上升,对这一持续问题进行持续评估至关重要。
与光滑念珠菌类似,热带念珠菌在世界不同地区的流行率差异很大。这种真菌在亚洲太平洋地区、加勒比地区和拉丁美洲尤为常见,但在北美和西欧则较少见。62 从历史上看,这种真菌对三唑类药物、棘白菌素类药物和两性霉素 B 均普遍敏感。然而,近年来对氟康唑和棘白菌素类药物耐药的报告日益增多,目前大多数地区对氟康唑的耐药基线水平为 3%至 5%,但在一些亚洲太平洋国家,这一比例高达 7%至 43%。115–117 大多数调查中对棘白菌素类药物的耐药率仍保持在 0%至 2%的较低水平。27,62 对两性霉素 B 耐药的情况仍然罕见,不过据印度的报告,泛耐药的热带假丝酵母菌可能占分离株的 1%。118
在导致侵袭性念珠菌病的五种最常见的念珠菌属物种中,近平滑念珠菌的致死率(全因死亡率)最低。18 根据地理区域的不同,它是导致侵袭性念珠菌病的第一至第四常见原因。18,62,66 然而,在过去十年中,氟康唑耐药性已出现,某些系列中超过 50% 的近平滑念珠菌分离株耐药。64,119 在美国,氟康唑耐药率在 4% 至 10% 之间;而在世界其他地区,包括日本、西欧和拉丁美洲,这一比例要高得多。尽管由于 FKS1 基因的自然多态性,其对棘白菌素类药物的敏感性最低限值(MIC)比其他念珠菌属物种高,但对棘白菌素类药物的耐药性仍相对少见(0% 至 3%)。62,103,112,120
克鲁斯念珠菌被认为对氟康唑固有耐药,但对伏立康唑和泊沙康唑的敏感性则有所不同。62,103,112 通常认为伏立康唑是治疗克鲁斯念珠菌感染的最佳口服药物选择,但最近的人群调查显示,伏立康唑的耐药率正在上升,在世界某些地区已接近 10%。棘白菌素和两性霉素 B 耐药性似乎也在出现,在较大的多中心研究中,耐药率在 3% 至 10% 之间。107,112
自 2009 年在全球范围内出现以来,耳念珠菌在临床领域相对较新。虽然在美国是引起侵袭性念珠菌病的较少见原因,但该菌在印度和南非是念珠菌血流分离株中最常见的。118,121 它以潜在的多重抗真菌耐药性而著称,但数据显示,70% 至 90% 的分离株对氟康唑耐药,且对季铵盐消毒剂不敏感,这使得该病原体能够在医疗相关表面持续存在并引发暴发。122,123 对棘白菌素(高达 7%)和两性霉素 B(高达 35%)的耐药性在不同地理区域差异很大。122–124 在南亚的印度,对所有三类抗真菌药物耐药的情况最为常见(3%),而在世界其他地区则少见得多。122–124 由于耳念珠菌存在人与人之间的传播可能性,因此感染预防措施对于防止医院内传播至关重要。尽管许多念珠菌属菌种具有非常可预测的抗真菌药物敏感性模式(例如白色念珠菌),但对所有具有临床意义的分离株进行种水平鉴定并随后进行抗真菌药物敏感性检测仍然至关重要。125 如果常规检测不可用,应定期对足够数量的本地分离株进行表征,以便能够充分预测特定医院或地理区域中该种属的敏感性模式。
IC 通用管理原则
美国感染病学会(IDSA)125 及欧洲指南126,127 建议,在未鉴定至菌种水平之前,对于大多数侵袭性念珠菌病患者(无论是否中性粒细胞减少),首选棘白菌素类药物作为初始治疗,因其具有广谱活性、良好的副作用特征、降低死亡率的优势,以及非白色念珠菌属中唑类耐药率不断上升的情况。125,128–132 两份指南均建议,若临床症状改善且药敏试验或菌种鉴定未提示三唑类耐药,则在 5 - 7 天(IDSA)或 10 天(欧洲指南)后降级为氟康唑治疗。由于克鲁斯念珠菌对氟康唑固有耐药,因此对于该菌种,指南推荐伏立康唑作为降级治疗药物。125,133
对于置有中心静脉导管的非中性粒细胞减少症患者,应拔除导管;但对于可能存在胃肠道念珠菌血症来源的中性粒细胞减少症患者,建议采取个体化的中心静脉导管拔除策略,但念珠菌近平滑念珠菌 IC.125 感染的情况除外。对于持续性念珠菌血症,建议进行超声心动图检查以评估是否存在心内膜炎。134 对于单纯性念珠菌血症,在血培养转阴且症状缓解后,治疗应持续 2 周。所有感染的心内装置均需移除或在疾病控制后进行无限期口服唑类药物抑制。125,132
由于潜在的眼科并发症会对治疗产生重大影响,美国感染病学会(IDSA)建议,对于非中性粒细胞减少症患者,应在确诊后一周内进行散瞳眼底检查;对于中性粒细胞减少症患者,则应在中性粒细胞减少症缓解后一周内进行检查,因为直到免疫功能恢复,才可能出现脉络膜视网膜炎和眼内炎的证据。根据 IDSA 和欧洲指南,对于眼部受累情况,应联合眼科进行玻璃体内治疗,若对氟康唑或伏立康唑敏感,则使用这两种药物。否则,推荐使用玻璃体内两性霉素 B,可联合氟胞嘧啶,必要时考虑进行玻璃体切除术。治疗持续 4 至 6 周,或直至眼科复查结果恢复正常。125,132
美国感染病学会(IDSA)建议,对于慢性播散性念珠菌病,应使用脂质体两性霉素 B 或棘白菌素类药物治疗数周,若对氟康唑敏感,则可逐步换用氟康唑,直至复查影像学显示病灶消退后方可停药。在高危化疗或干细胞移植期间,建议持续治疗以预防复发。
对于骨髓炎,若出现大脓肿或关节不稳定,则需进行外科清创术。美国感染病学会(IDSA)建议使用口服唑类药物治疗 6 至 12 个月,或使用棘白菌素类药物(较弱的替代药物为脂质体两性霉素 B)治疗 2 周,随后改为口服唑类药物治疗 6 至 12 个月。欧洲指南倾向于使用口服唑类药物治疗 6 至 12 个月,或使用脂质体两性霉素 B 治疗 2 至 6 周,随后改为口服唑类药物完成 6 至 12 个月的疗程。对于化脓性关节炎,建议进行外科引流并移除任何人工装置,若装置保留,则需长期口服唑类药物进行抑制。IDSA 建议使用口服唑类药物治疗 6 周,或使用棘白菌素类药物治疗 2 周,随后使用口服唑类药物治疗≥4 周,或使用脂质体两性霉素 B 治疗 2 周,随后改为口服唑类药物治疗≥4 周。欧洲指南建议口服唑类药物治疗 6 周以上,或使用脂质体两性霉素 B 治疗 2 周,随后改为氟康唑治疗 4 周以上,伏立康唑治疗 6 周以上也可作为替代方案。125,132
中枢神经系统(CNS)念珠菌病的最佳治疗方案数据较少。美国感染病学会(IDSA)建议使用脂质体两性霉素 B 联合或不联合氟胞嘧啶进行治疗,若临床反应良好且对氟康唑敏感,可考虑降阶梯治疗。治疗时长取决于症状、影像学及脑脊液异常的消退情况。正常剂量下棘白菌素类药物对中枢神经系统的渗透性较差,不推荐使用。125 欧洲指南弱推荐静脉注射脂质体两性霉素 B 联合氟胞嘧啶治疗 6 周,随后使用氟康唑治疗 3 周,或者使用脂质体两性霉素 B 联合氟康唑治疗 4 周,具体取决于药敏情况。132
早期治疗的必要性
侵袭性念珠菌病(IC)的归因死亡率仍然很高,且随着治疗的延迟而增加。念珠菌血培养平均需要 2 至 3 天才能生长,具体时间取决于菌种。因此,预防和经验性治疗策略均被常规采用以降低死亡率。133,135,136 IDSA 建议在侵袭性念珠菌病发生率超过 5%的重症监护病房(ICU)中,对高危患者考虑使用氟康唑或棘白菌素类药物进行预防。125,137一项针对非中性粒细胞减少性重症监护病房(ICU)患者预防措施的 Cochrane 综述未发现对死亡率有影响,但确实指出侵袭性真菌感染显著减少。138 欧洲指南建议对近期腹部手术后出现反复穿孔或渗漏的 ICU 患者使用氟康唑进行预防。132 对于异基因干细胞移植患者,在初始中性粒细胞减少期,推荐使用氟康唑、泊沙康唑、伏立康唑或米卡芬净进行预防,随后根据免疫恢复情况和移植物抗宿主病的免疫抑制情况,转为使用氟康唑或泊沙康唑。131
美国感染病学会(IDSA)建议,对于有风险因素和感染替代标志物且病情恶化的患者,应考虑经验性抗真菌治疗。在出现感染性休克的情况下,应尽快开始治疗。不过,对于感染性休克且有侵袭性念珠菌病风险因素的患者,经验性抗真菌治疗的益处存在争议。EMPIRICUS 试验未能证明在有感染性休克和侵袭性念珠菌病风险因素的 ICU 患者中使用经验性米卡芬净的益处。139
侵袭性念珠菌病的抗真菌药物研发管线尽管侵袭性念珠菌感染相关的发病率和死亡率仍然很高,但目前仅批准了四类主要的抗真菌药物用于全身治疗。棘白菌素类药物作为大多数念珠菌感染的一线治疗药物,在临床试验中侵袭性念珠菌病的总体失败率为 25% 至 30%,并且在泌尿道内几乎没有活性。18,133,140–144 由于多种机制(表 2),多重耐药念珠菌感染呈上升趋势,尤其是耳念珠菌,其对唑类药物广泛耐药,并且在治疗期间有可能对所有类别的抗真菌药物产生耐药性,这凸显了开发新药以应对流行病学变化的必要性。133,145–153
表 2. 主要念珠菌属物种最常见的耐药机制念珠菌物种
抵抗机制C. albicans白色念珠菌
氟康唑——排出转运蛋白(MDR1、CDR1、CDR2),其氨基酸替代较少见C. glabrata
光滑念珠菌
阿唑类——药物排出体(Cdr1和Cdr2)的变化
嵌棘白素——葡聚糖合酶基因中的FKS1多态性
突变表型——不匹配修复缺陷导致多种抗真菌耐药性表型C. krusei
克柔假丝酵母菌
氟康唑——由于ERG11内部发生变化而具有内在耐药性,其他唑类常被保留C. parapsilosis假丝酵母菌
氟康唑——ERG11中氨基酸取代(主要是Y132F),最常见的是对伏立康唑的敏感性不一C. lusitaniae
葡萄牙假丝酵母
两性霉素B——人工麦角甾醇合成基因(ERG3等)的突变或表达改变。C. auris(耳念珠菌)
阿唑类——大多数为氟康唑耐药性(ERG11突变),且对其他唑类有不同易感性
两性霉素B——假设由麦角甾醇生物合成的变化引起棘棘白素——由FKS1突变引起
令人鼓舞的是,2023 年美国食品药品监督管理局批准了一种新的棘白菌素类药物——瑞扎芬净(rezafungin),用于治疗成人侵袭性真菌感染。在一项名为 ReSTORE 的 3 期临床试验中,瑞扎芬净与卡泊芬净(caspofungin)治疗侵袭性念珠菌病的疗效相当,治愈率达 60%。154,155 它的结构与阿尼芬净(anidulafungin)相似,但半衰期更长,可每周给药一次,这将使其更适合门诊治疗。149 目前,包括 ReSPECT 在内的其他研究正在进行中,ReSPECT 是一项针对接受异基因造血干细胞移植患者预防侵袭性真菌感染的 3 期临床试验。150,156
伊布替芬净(Ibrexafungerp)是另一种新型药物,它在与棘白菌素不同的位点抑制(1→3)-β-D-葡聚糖合成,因此对许多因 FKS 突变而对棘白菌素耐药的菌株仍具有活性,包括许多耳念珠菌菌株。156 重要的是,它在酸性环境中仍具有溶解性和活性,使其适用于脓肿治疗。149,157 针对其他药物治疗无效的感染的临床试验结果令人鼓舞。150伊布替芬的口服生物利用度为 35% 至 50%,全身分布良好,但中枢神经系统除外。目前,伊布替芬仅获美国食品药品监督管理局批准用于治疗外阴阴道念珠菌病,不过针对侵袭性念珠菌病降阶梯治疗、难治性念珠菌病以及耳念珠菌的临床试验正在进行中。149,156–159
最后,福沙匹坦(fosmanogepix)是另一种令人鼓舞的新型抗真菌药物。福沙匹坦是一种首创的 Gwt1 真菌蛋白抑制剂,Gwt1 蛋白类是将甘露糖蛋白锚定在细胞膜和细胞壁所必需的。160 这种新型机制对包括耐药念珠菌在内的多种念珠菌属具有广泛的活性。160 一项针对耳念珠菌所致念珠菌血症患者的 2 期临床试验结果表明,福沙匹坦安全有效。161 此外,一项针对由白色念珠菌、光滑念珠菌、近平滑念珠菌和/或都柏林念珠菌所致念珠菌血症患者的 2 期研究数据显示,20 名患者中有 16 名成功治愈,且未出现严重不良事件。162 3 期试验正在计划中,但鉴于抗真菌耐药率不断上升,这些初步结果令人鼓舞。
医疗环境中的预防与感染控制
鉴于与导管相关血流感染(CLABSI)相关的发病率和死亡率,预防是关键的优先事项。为此,美国感染病学会(IDSA)合作制定了多项干预措施的联合建议,以降低所有 CLABSI(包括念珠菌血症)的风险。163 这些 CLABSI 防护包的实施降低了感染的发生率。18 指南包括对置管适当指征的教育,指定特定人员并采用无菌技术来置管和护理中心静脉导管。上肢部位的感染风险低于下肢部位。163 所有置管部位应每日进行评估,如果出现静脉炎、感染或功能障碍的迹象,应移除外周导管。163 非隧道式锁骨下导管的感染风险低于颈内静脉或股静脉部位,建议使用最少数量的管腔。163 及时移除不必要的中心静脉导管可降低感染风险,所有置管和中心静脉导管的使用都应采用无菌技术和无菌屏障预防措施。163在置管前应进行氯己定皮肤清洁,根据敷料材质制定无菌敷料更换计划。只有在导管出现故障或怀疑感染时才更换中心静脉导管和中线导管,常规更换并不能降低感染率。163 对重症监护病房患者进行每日氯己定沐浴可降低包括念珠菌血症在内的血流感染风险。125,164
医疗机构中耳念珠菌的暴发近期有所增加,这在很大程度上是由新冠疫情造成的,大量危重症患者需要机械通气,且感染控制措施可能出现了漏洞。145 该病原体在医疗机构环境中可长期存在,患者可能在一年以上仍处于定植状态。由于耳念珠菌在医疗机构中具有持续存在和传播的能力,美国疾病控制与预防中心发布了相关指南以限制其暴发。165,166 定植筛查是预防医疗机构中耳念珠菌传播的关键策略,通过识别未被察觉的定植者,从而采取适当的感染预防和控制措施。确定筛查对象取决于诸多因素,但应考虑对有高风险感染耳念珠菌的个体进行筛查,包括有流行病学关联的病例、在高风险机构有过接触史以及存在相关风险因素的患者。建议使用双侧腋窝和腹股沟的复合拭子进行筛查。感染耳念珠菌的患者应采取适当的隔离预防措施,这包括在急性护理医院采取接触预防措施,但在养老院环境中可考虑采取加强的屏障预防措施。由于患者通常会长期携带耳念珠菌,且检测结果可能时而呈阴性,因此在住院期间以及未来住院期间应持续采取隔离预防措施,不建议重复筛查以“清除”患者从而解除隔离预防措施。鉴于耳念珠菌在环境中的顽强生存能力,建议每天至少使用适当的消毒剂对感染患者的房间以及他们接触过的任何表面或共用设备(如血糖仪、移位机、体温计等)进行清洁。
结论
侵袭性念珠菌病仍然是全球性的威胁。由于全球监测有限,再加上诊断方面的挑战,即便有新的临床诊断方法,该问题的严重程度可能仍被低估。尽管预防策略有所进步,但侵袭性念珠菌病仍是代价高昂且致病严重的疾病。虽然总体发病率似乎在下降,但治疗选择和感染控制措施对全球和局部的流行病学情况有着重大影响。非白色念珠菌属的增多以及多重耐药病原体 C. auris 的出现,将在不断演变的治疗实践中发挥重要作用,并影响该疾病的未来流行病学情况。尽管近期在治疗方面取得了进展,但鉴于突破性感染和耐药性的增加,仅靠抗真菌治疗在没有全面的念珠菌感染预防策略的情况下,不太可能成为有效的解决方案。需要在预防策略方面取得进展,并开发新的药物,以降低全球发病率并改善治疗效果。
参考文献(略)。
欢迎指正错误和交流。
若光医学中心诺光门诊部(长沙)预约(下图)。
中国女性洁净计划
下图中二维码,可以扫码购买。、
真菌专题(目录)
(1)全球范围内抗真菌药物耐药性的出现对人类健康和粮食安全构成挑战
(2)关于《微生物分子生物学评论》(MMBR)中有关真菌性行为的专题集:分子机制与进化意义
(3)世界卫生组织真菌优先病原体清单成为游戏规则的改变者
(4)针对世界卫生组织重点真菌病原体的新型治疗选择
(5)耳念珠菌的遗传学与出现
(6)全球严重真菌病的发病率和死亡率
(7)在透析单位管理耳念珠菌:挑战、策略及未来方向
(8)医疗机构中预防耳念珠菌传播的策略
本文:(9)侵袭性念珠菌病的流行病学
(10)耳念珠菌中心静脉导管相关血流感染在危重症患者中的情况:糟糕局面中的最坏情况
(11)耳念珠菌感染
(12)耳念珠菌:宿主相互作用、抗真菌药物耐药性及诊断
(13)念珠菌病诊断与管理全球指南:欧洲临床微生物学与传染病学会(ECMM)与国际真菌病学会(ISHAM)及美国微生物学会(ASM)合作倡议
(14)多重耐药念珠菌:流行病学、分子机制及治疗
(15)抗真菌药物耐药性:白色念珠菌及其他物种中的分子机制
(16)耳念珠菌:一种近期出现的多重耐药性真菌病原体
(17)真菌细胞壁生物发生:结构复杂性、调控及抑制
(18)单基因突变是新兴真菌病原体耳念珠菌代谢适应和获得丝状体能力的基础
(19)Gcn5 赖氨酸乙酰转移酶介导耳念珠菌的细胞壁重塑、抗真菌药物耐药性以及毒力
(20)抗 Hyr1p 单克隆抗体对多重耐药耳念珠菌所致系统性念珠菌病的保护效力
(21)在“同一健康”的背景下理解抗真菌耐药性的临床和环境驱动因素
(22)奥特塞康唑(Oteseconazole):复发性外阴阴道念珠菌病治疗的新进展
(23)奥特塞康唑与氟康唑治疗严重外阴阴道念珠菌病的疗效比较:一项多中心、随机、双盲、Ⅲ期临床试验
(24)奥特塞康唑:期待已久的抗真菌药物新成员,为复发性外阴阴道念珠菌病(RVVC)的治疗带来新选择
(25)氟胞嘧啶及其临床应用
(26)耳念珠菌会经历依赖黏附素和不依赖黏附素的细胞聚集
(27)耳念珠菌中由 ACE2 缺失介导的细胞聚集调节皮肤真菌定植和宿主免疫反应
(28)新出现的真菌病原体耳念珠菌诱导干扰素γ定植于哺乳动物毛囊
(29)在“同一健康”的背景下理解抗真菌耐药性的临床和环境驱动因素
(30)瑞扎芬净(瑞扎约)用于成人念珠菌血症和侵袭性念珠菌病治疗的审批中的监管考量
(31)在治疗过程中体内出现高水平耐药性揭示了耳念珠菌对两性霉素 B 耐药性的首个已知机制
(32)耳念珠菌细胞表面黏附素的功能冗余对于细胞间相互作用和聚集至关重要
(33)新出现的真菌病原体耳念珠菌诱导IFNγ 以定植皮肤
(34)干扰素-γ在自身免疫性多内分泌腺综合征 1 型中的作用
(35)针对耳念珠菌这种真菌病原体的先天免疫反应
(36)调控耳念珠菌病理生物学特征及抗真菌药物耐药性的信号通路
(37)人体真菌病原体的免疫反应及治疗前景
(38)针对耳念珠菌这种真菌病原体的先天免疫反应
(39)耳念珠菌对氟胞嘧啶的体外快速耐药性进化
(40)全基因组分析实验进化而来的耳念珠菌揭示了多种新型多重耐药机制
(41)黏附素 Als4112 通过与角质形成细胞和细胞外基质蛋白的相互作用促进耳念珠菌皮肤定植
(42)全球范围内抗真菌药物耐药性的出现对人类健康和粮食安全构成挑战
(43)白色-棕色转换控制耳念珠菌的表型可塑性和毒力
(44)抗真菌药物耐药性的分子机制
(45)美国真菌病的经济负担
~~等。
微生物疗法临床研究
2025-12-30
引言:十年期待与现实落差的拷问
2015年,首项分子POCT检测获得CLIA豁免,行业曾预言这将掀起诊断领域的颠覆性变革——让核酸检测摆脱专业实验室束缚,在床旁、基层实现"即测即得",既取代灵敏度不足的快速免疫测定,又弥补传统实验室检测的时效短板。然而十年过去,全球已涌现180款分子POCT设备、94款通过监管审查,却仍面临"技术热、市场冷"的尴尬:多数产品装机量低迷,部分企业折戟沉沙,甚至有人质疑其"上量难"的背后是需求本身的虚假。分子POCT究竟是临床急需的"潜力股",还是资本催生的"伪需求"?答案藏在技术演进、临床痛点与市场生态的三重博弈中。
一、真需求的铁证:无法替代的临床价值1. 急症场景的"时间战场"
急诊感染、重症监护中,每一分钟都关乎预后。传统实验室检测需数小时出结果,而分子POCT能将这一时间压缩至15-30分钟——生物梅里埃SpotFire系统15分钟完成呼吸道病原体检测,卡尤迪Flash10更是实现20分钟出结果,让医生在患者就诊期间即可制定治疗方案,避免无效用药和病情延误。亨利·福特医疗系统引入罗氏Liat系统后,其新冠疫情应对效率大幅提升,至今仍在流感快速检测中发挥核心作用,印证了急症场景的刚性需求。2. 基层与特殊场景的"诊断补位"
在资源匮乏的农村地区,分子POCT可实现结核病、淋病等疾病的"检测-治疗"闭环,既挽救生命又遏制超级细菌传播;在入境口岸、社区诊所等非实验室场景,其小巧便携、操作简便的特性,让核酸检测触达传统手段难以覆盖的角落。卡尤迪Flash10无需专业实验室分区,插电即可使用,真正实现技术下沉,解决了基层"有设备不会用、会用没设备"的困境。3. 精准医疗的"决策支撑"
相比传统检测,分子POCT兼具高灵敏度与多重检测能力——国内某企业产品可同时检测16种病原体,SpotFire提供5靶标小面板与14靶标大面板组合,能精准区分复杂感染中的致病元凶,避免抗生素滥用。北京协和医院赵颖研究员指出,这种"单一样本多病原体检测"能力,为循证医学提供了关键依据,是感染性疾病、肿瘤基因检测等领域的重要支撑。二、上量难的症结:需求真实,障碍多重1. 成本与报销的"双重挤压"
分子POCT设备与试剂的高成本的是核心壁垒。尽管临床价值显著,但医保报销机制未能同步跟进,导致成本多转嫁给医疗机构或患者,限制了大规模采购。美国作为成熟市场,直到2024年才为SpotFire呼吸道检测设立专用报销代码,其装机量随即从1200台跃升至4000台,印证了报销政策对市场放量的关键作用。2. 临床指南与认知的"循环困境"
缺乏明确的临床效用研究和指南支持,让医疗机构不敢贸然大规模采用——美国A组链球菌分子POCT已获批十年,但传染病学会指南仍停留在2012年,要求培养确认,直接制约其推广。而没有广泛应用,就难以积累足够数据支撑指南更新,形成"无指南→低采用→无数据→无指南"的恶性循环。3. 技术落地与生态的"适配短板"
部分产品存在"技术先进,落地脱节"问题:早期低通量系统难以应对病例激增,小型企业缺乏大规模制造能力;部分产品操作流程与现有医疗 workflows不兼容,需要额外人员培训和质量管控流程,增加了医疗机构的隐性成本。赛沛前CEO约翰·毕晓普直言,行业缺乏"推动技术落地的愿景与行动力",而非技术本身存在缺陷。4. 市场教育与患者认知的"滞后性"
后新冠时代,患者对即时结果的期望显著提升,但多数医疗机构与患者对分子POCT的认知仍停留在"新冠检测"层面,对其在流感、呼吸道感染、性传播疾病等领域的应用价值了解不足。缺乏市场教育导致需求端启动缓慢,难以形成规模化采购效应。
三、破局信号:从技术突围到市场爆发1. 技术迭代破解核心痛点
第四代iPOCT技术融合互联网与自动化,正在突破传统瓶颈:卡尤迪FlashDetect Robo采用智能机械臂加样,24小时可处理2300个样本,兼顾快速检测与批量处理能力;超多重检测技术实现"一盘多检",进一步提升单检测的性价比。CRISPR、石墨烯生物传感器等新技术的应用,更让检测成本持续下降,为上量奠定基础。2. 标杆企业验证放量路径
生物梅里埃通过"收购-整合-创新"模式,为行业提供了可复制的成功案例:收购BioFire获得极致PCR技术,拿下CLIA豁免与医保报销代码,拓展检测菜单至心脏标志物等领域,最终实现SpotFire系统172%的销售额增长,2025年预计收入达1.7亿欧元。其成功关键在于精准定位急诊场景、打通报销环节、持续扩展应用边界。3. 政策与需求形成共振
全球医疗系统对感染性疾病防控、抗微生物药物管理的重视,推动分子POCT成为刚需。美国FDA已为11款产品授予510(k)许可,国内多款产品获得NMPA与CE双重认证,监管环境持续优化。随着临床数据积累,指南更新已箭在弦上,有望打破长期制约市场的认知壁垒。结语:伪需求争议落幕,真刚需加速破局
分子POCT的"上量难",从来不是需求本身的虚假,而是技术、政策、市场生态尚未完成协同的阶段性问题。从临床场景的刚性需求来看,它解决了传统诊断"慢、远、繁"的核心痛点;从技术演进来看,自动化、多重化、低成本化正在破解落地障碍;从市场信号来看,标杆企业的爆发已验证其商业潜力。
随着报销政策完善、临床指南更新、技术成本下降,分子POCT将从"小众选择"变为"主流标配",伪需求的争议终将落幕。对于行业而言,当下的关键不是质疑需求,而是聚焦"如何让好技术被广泛使用"——这需要企业深耕临床价值、政策层面优化支持、行业共同做好市场教育。当技术创新与临床需求、市场生态形成合力,分子POCT的放量增长将水到渠成。
文章仅作分享,如有侵权请联系我们
点击上方蓝色文字关注我们
诊断试剂
100 项与 Biofire Diagnostics LLC 相关的药物交易
登录后查看更多信息
100 项与 Biofire Diagnostics LLC 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月22日管线快照
无数据报导
登录后保持更新
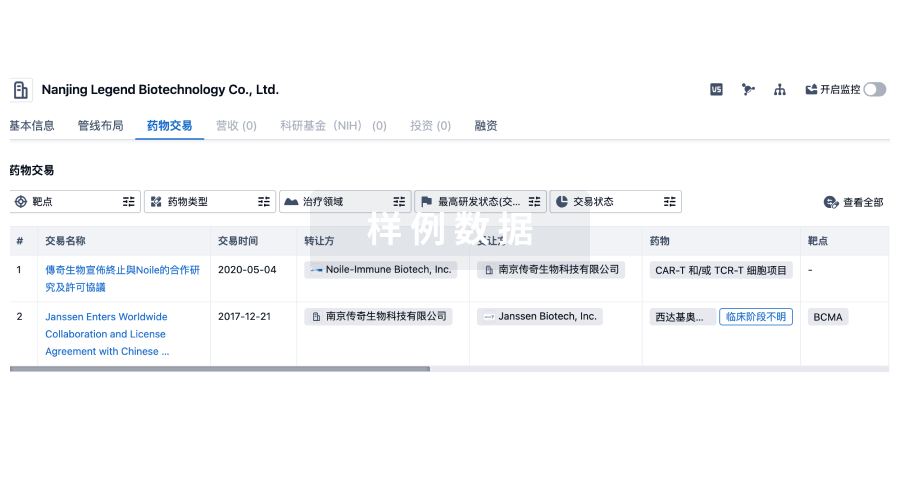
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
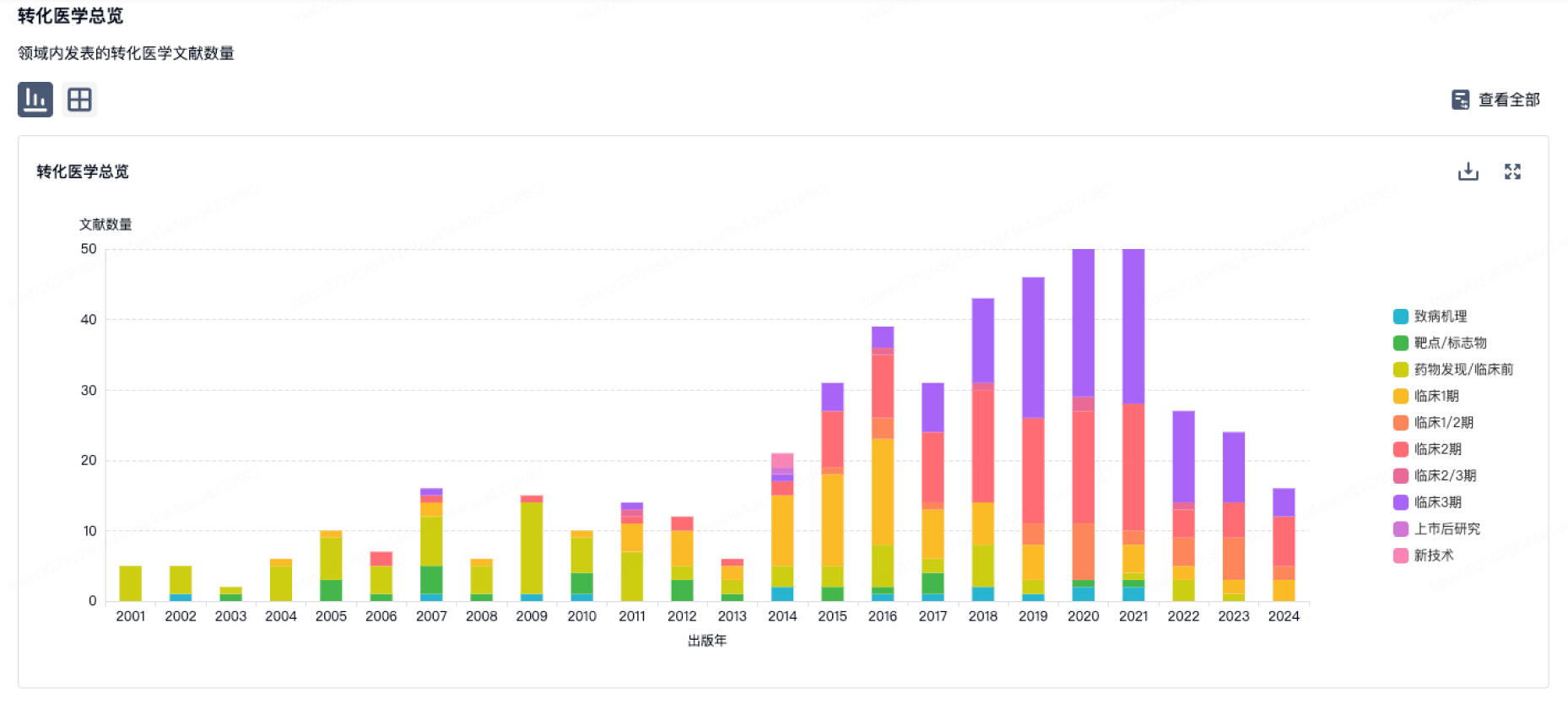
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用