预约演示
更新于:2026-04-12

Reata Pharmaceuticals, Inc.
更新于:2026-04-12
概览
标签
肿瘤
呼吸系统疾病
小分子化药
疾病领域得分
一眼洞穿机构专注的疾病领域

技术平台
公司药物应用最多的技术
靶点
公司最常开发的靶点
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
NCT05895552
A Phase 2 Study to Evaluate the Safety and Efficacy of RTA 901 in Patients With Diabetic Peripheral Neuropathic Pain
NCT04494646
BARCONA: A Phase II, Randomized, Double-blind, Placebo-controlled, Multi-center Study of the Effects of Bardoxolone Methyl in Participants With SARS-Corona Virus-2 (COVID-19)

NCT03366337
A Phase 2 Trial of the Safety and Efficacy of Bardoxolone Methyl in Patients With Rare Chronic Kidney Diseases
100 项与 Reata Pharmaceuticals, Inc. 相关的临床结果
登录后查看更多信息
登录后查看更多信息
2025-08-01JOURNAL OF THE NEUROLOGICAL SCIENCES
The clinical burden of Friedreich ataxia in the United States: A retrospective claims database analysis
Article
作者: Jiang, Anya ; Boudreau, Julien ; Lynch, David R ; Bajaj, Abhishek ; Setyawan, Juliana ; Hua, Qi ; Perlman, Susan ; Lawson, Richard ; Khan, Seemi ; Zhang, Su ; Yang, Hongbo
BACKGROUND:
Friedreich ataxia (FRDA) is a terminal, rare, autosomal recessive genetic disorder associated with debilitating comorbidities. However, limited real-world data on clinical outcomes associated with FRDA have been presented.
METHODS:
The Komodo Research Data (01/2016-03/2023) was used to identify patients with FRDA who had ≥2 independent non-diagnostic early-onset cerebellar ataxia codes, 1 non-diagnostic FRDA specific code, and ≥12 months of continuous enrollment before and after the index date (defined as a random date with FRDA diagnosis). Individuals without early-onset cerebellar ataxia were eligible for controls. The controls were matched at a 5:1 ratio on age, sex, insurance type, US region, and continuous enrollment and assigned the same index date as the matched patients.
RESULTS:
After matching, the study included 652 patients with FRDA and 3260 matched controls (mean age 33.2 years; 51.4 % females). During the follow-up period (median 26.2 months for cases and 28.3 months for controls), the incremental clinical burden in FRDA cases vs. matched controls was high: patients with FRDA had significantly higher odds of loss of ambulation (odds ratio: 158.0 [95 % confidence interval (CI): 112.4-222.3]), cardiomyopathy (59.2 [41.6-84.1]), scoliosis (49.0 [35.4-67.9]), falls (7.4 [5.9-9.2]), diabetes (2.5 [2.0-3.2]), head injury (2.4 [1.9-3.0]), and fracture (3.3 [2.6-4.2]) compared to controls (all p < 0.001). Patients with FRDA also experienced a higher risk of mortality than controls (hazard ratio: 3.9 [95 % CI: 2.4-6.4]).
CONCLUSIONS:
Real-world data suggested patients with FRDA were associated with substantial and incremental clinical burden compared to matched controls and will benefit in advances in treatment for FRDA.
2024-10-01CPT-Pharmacometrics & Systems Pharmacology
Understanding the mechanisms of food effect on omaveloxolone pharmacokinetics through physiologically based biopharmaceutics modeling
Article
作者: Hynes, Scott M. ; Walker, Deborah ; Pepin, Xavier J. H. ; Suarez‐Sharp, Sandra ; Semmens, Lois Q. ; Zahir, Hamim
Abstract:
Omaveloxolone is a nuclear factor (erythroid‐derived 2)‐like 2 activator approved in the United States and the European Union for the treatment of patients with Friedreich ataxia aged ≥16 years, with a recommended dosage of 150 mg orally once daily on an empty stomach. The effect of the US Food and Drug Administration (FDA) high‐fat breakfast on the pharmacokinetic profile of omaveloxolone observed in study 408‐C‐1703 (NCT03664453) deviated from the usual linear correlation between fed/fasted maximum plasma concentration (Cmax) and area under the concentration–time curve (AUC) ratios reported for various oral drugs across 323 food effect studies. Here, physiologically based biopharmaceutics modeling (PBBM) was implemented to predict and explain the effect of the FDA high‐fat breakfast on a 150‐mg dose of omaveloxolone. The model was developed and validated based on dissolution and pharmacokinetic data available across dose‐ranging, food effect, and drug–drug interaction clinical studies. PBBM predictions support clinical observations of the unique effect of a high‐fat meal on omaveloxolone pharmacokinetic profile, in which the Cmax increased by 350% with only a 15% increase in the AUC. Key parameters influencing omaveloxolone pharmacokinetics in the fasted state based on a parameter sensitivity analysis included bile salt solubilization, CYP3A4 activity, drug substance particle size distribution, and permeability. Mechanistically, in vivo omaveloxolone absorption was solubility and dissolution rate limited. However, in the fed state, higher bile salt solubilization led to more rapid dissolution with predominant absorption in the upper gastrointestinal tract, resulting in increased susceptibility to first‐pass gut extraction; this accounts for the lack of correlation between Cmax and AUC for omaveloxolone.
2024-04-01Bioanalysis
Development of LC–MS/MS Method for Cyanoenone Triterpenoid Determination to Support CNS Tissue Distribution Study
Article
作者: Tian, Lynn ; Tamer, Edward ; Tian, Qingguo ; Lafon, William
Aims: Cyanoenone triterpenoids penetrate the CNS, exhibiting biological activity via the nuclear factor E2-related factor (Nrf2) pathway. This is the first report on methods for the quantification of cyanonenone triterpenoids' distribution in various CNS tissues by LC-MS/MS. Materials & methods: The analyte was extracted from brain tissue homogenate using protein precipitation and supported liquid extraction. Results & conclusion: The assay validated a quantification range of 3.00-3000 ng/g in brain tissue samples as low as 5 mg. All parameters, including interference (≤20% at LLOQ) and accuracy/precision (15%, with 20% at LLOQ), met acceptance criteria. This assay supported a CNS distribution study, analyzing more than 10 mouse brain regions successfully.
2026-04-01
2026-03-31
100 项与 Reata Pharmaceuticals, Inc. 相关的药物交易
登录后查看更多信息
100 项与 Reata Pharmaceuticals, Inc. 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年06月24日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
临床前
1
9
其他
登录后查看更多信息
当前项目
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
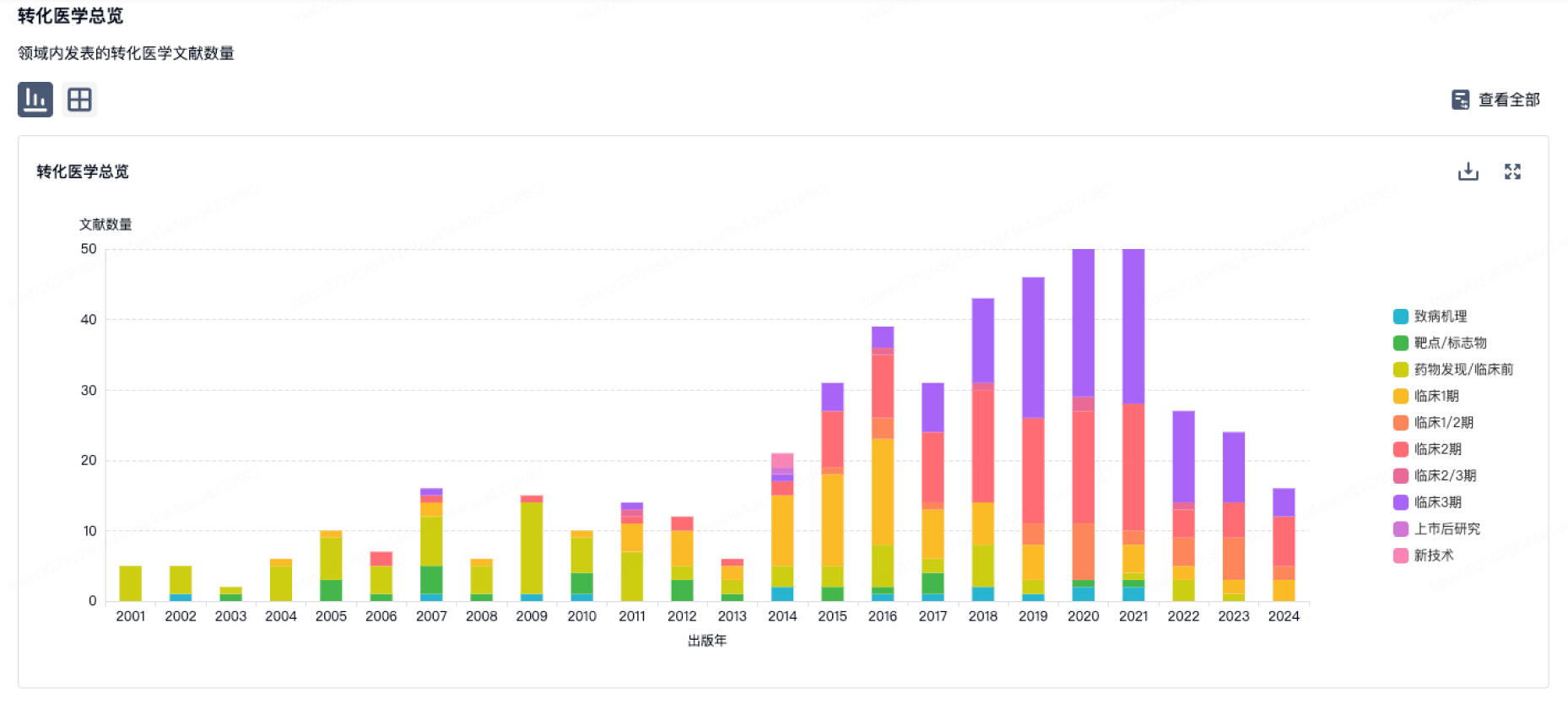
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
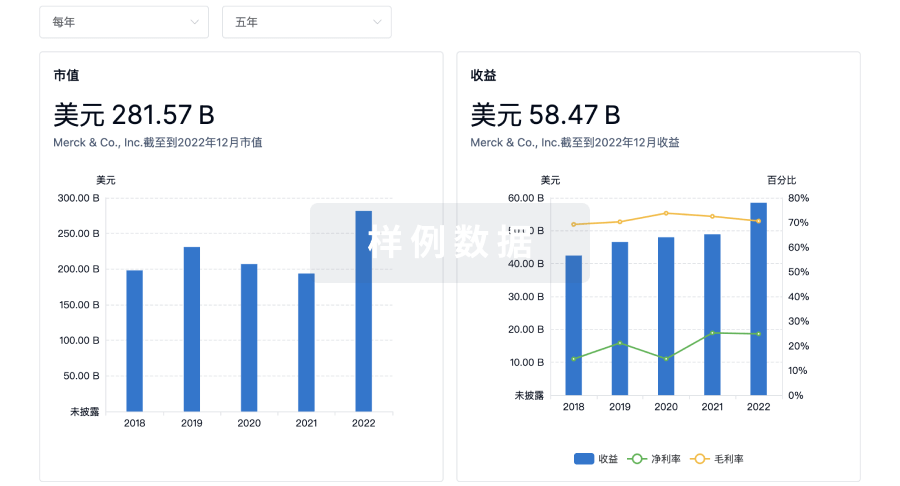
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
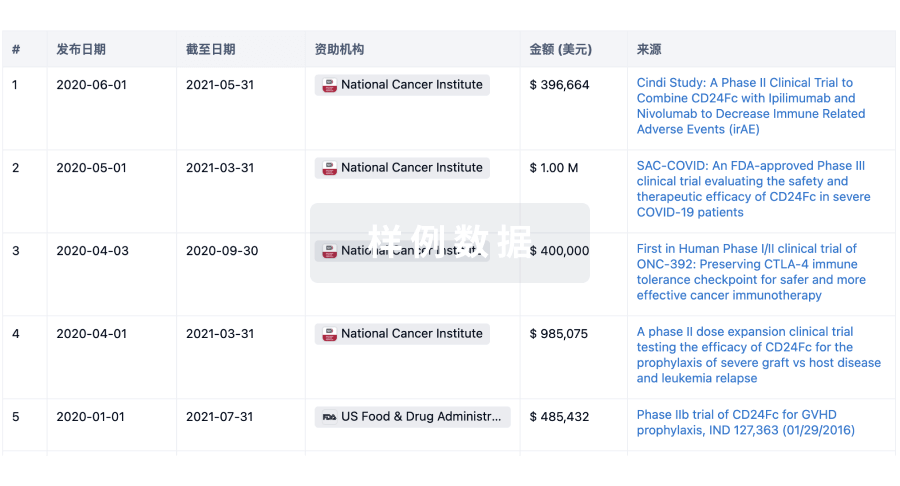
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用