预约演示
更新于:2025-12-09

Structural Genomics Consortium
更新于:2025-12-09
概览
标签
感染
小分子化药
疾病领域得分
一眼洞穿机构专注的疾病领域

技术平台
公司药物应用最多的技术
靶点
公司最常开发的靶点
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
100 项与 Structural Genomics Consortium 相关的临床结果
登录后查看更多信息
登录后查看更多信息
2025-10-23JOURNAL OF MEDICINAL CHEMISTRY
Enabling Open Machine Learning of Deoxyribonucleic Acid-Encoded Library Selections to Accelerate the Discovery of Small Molecule Protein Binders
Article
作者: Harding, Rachel J. ; Lengyel-Zhand, Zsofia ; Zhu, Hongyao ; Joseph, Jermiah ; Klug-McLeod, Jacquelyn L. ; Bagale, Nabin ; Couñago, Rafael M. ; Bolotokova, Albina ; Montgomery, Justin I. ; Owen, Dafydd R. ; Liu, Jian ; Reza, Shaghayegh ; Cheng, Xuemin ; Harris, Anthony R. ; Ghiabi, Pegah ; Tu, Guiping ; Halabelian, Levon ; Zhang, Jinzhi ; Zeng, Hong ; Dong, Aiping ; Edwards, Aled M. ; Haibe-Kains, Benjamin ; Lee, Jisun ; Ahmad, Shabbir ; Gibson, Elisa ; Tropsha, Alexander ; Foley, Timothy L. ; Li, Xianyang ; Sakata, Sylvie ; Wellnitz, James ; Dou, Dengfeng ; Li, Jin
Machine learning (ML) is increasingly used in DNA-encoded library (DEL) screening for ligand discovery, but its success depends on access to suitable data sets, which are often proprietary and costly. To overcome this, we present the first fully open, automated DEL-ML framework using public DEL data sets and chemical fingerprints to enable reproducible, accessible drug discovery. Our workflow─from model training to virtual screening and compound selection─requires no human intervention. As a proof of concept, we identified binders for WDR91 by training ML models on the HitGen OpenDEL library (3B molecules) and screening the Enamine REAL Space library (37B molecules), yielding 50 candidates. Experimental testing confirmed seven novel binders with dissociation constants between 2.7-21 μM. Our open-source approach matches the performance of proprietary methods, demonstrating that public DEL data can support robust ML-driven ligand discovery and fostering transparency and broader community participation in drug development.
2025-10-01JOURNAL OF THE AMERICAN SOCIETY FOR MASS SPECTROMETRY
Site-Specific Hydrogen Deuterium Exchange Difference Mass Spectrometry Measurements for Ligand Binding
Article
作者: Ackloo, Suzanne ; Arrowsmith, Cheryl ; Leblanc, Yves ; Wilson, Derek J. ; Anacleto, Joseph ; Wolf, Esther ; Lento, Cristina
Conventional bottom-up HDX-MS experiments are highly suitable for use in drug development; however, a major limitation of this approach is that it generally provides only peptide-level structural resolution. Site specific (i.e., single amino acid-resolved) HDX-MS measurements have been achieved using ECD/ETD, but the low efficiency of these fragmentation techniques, combined with poor ion transmission associated with 'detuning' the instrument to fully prevent deuterium scrambling, results in sensitivity losses that make ligand binding measurements impractical in a 'real-world' (e.g., drug development) context. Here we apply a recently developed method for zero scrambling, high efficiency ECD in the challenging context of ligand binding differential HDX experiments, demonstrating a wealth of additional information that can be acquired when HDX-MS analyses are conducted at the amino acid level.
2025-08-08ACS Infectious Diseases
Enantioselective Chemical Probe for Chikungunya nsP2 Helicase with Antialphaviral Activity
Article
作者: Hossain, Mohammed Anwar ; Talbot, Kacey M. ; Chen, Chun-Hsing ; Hirsch, Alec J. ; Garcia Perez, Julia ; Muthu Ramalingam, Bose ; Arrowsmith, Cheryl H. ; Heise, Mark T. ; Willson, Timothy M. ; Houliston, Scott ; Vala, Anand ; Amare, Meareg G. ; Moorman, Nathaniel J. ; Brown, Peter J. ; Cameron, Craig E. ; Oh, Hans J. ; Couñago, Rafael M. ; Li, Fengling ; Vargason, Ava ; Smith, Jessica L. ; Sears, John D. ; Halfmann, Peter ; Liu, Shubin ; Arnold, Jamie J. ; Halabelian, Levon
Chikungunya virus (CHIKV) replication relies on the multifunctional nsP2 protein, making it an attractive target for antiviral drug discovery. Here, we report the resolution of oxaspiropiperidine 1, a first-in-class inhibitor of the CHIKV nsP2 RNA helicase (nsP2hel), into its constitutive enantiomers and characterization of their antiviral activity. The enantiomer (R)-1 exhibited potent inhibition of viral replication, nsP2hel ATPase activity, and dsRNA unwinding, while the (S)-1 enantiomer was >100-fold less active. The (R)-1 enantiomer also demonstrated a high selectivity for CHIKV over other RNA viruses and for nsP2hel over other RNA helicases. Direct binding of (R)-1 to the nsP2hel protein was confirmed by 19F NMR. Biophysical and structural studies revealed conformational polymorphism in the spirocyclic scaffold of (R)-1, suggesting a potential role of thermal mobility of the ligand in allosteric inhibition of nsP2hel. Collectively, these findings designate (R)-1 (RA-NSP2-1) as a high-quality chemical probe and (S)-1 (RA-NSP2-1N) as a negative control for probing the biology of alphavirus RNA helicases.
2024-04-25
·美通社
100 项与 Structural Genomics Consortium 相关的药物交易
登录后查看更多信息
100 项与 Structural Genomics Consortium 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2026年05月21日管线快照
管线布局中药物为当前组织机构及其子机构作为药物机构进行统计,早期临床1期并入临床1期,临床1/2期并入临床2期,临床2/3期并入临床3期
药物发现
1
1
临床前
其他
1
登录后查看更多信息
当前项目
登录后查看更多信息
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
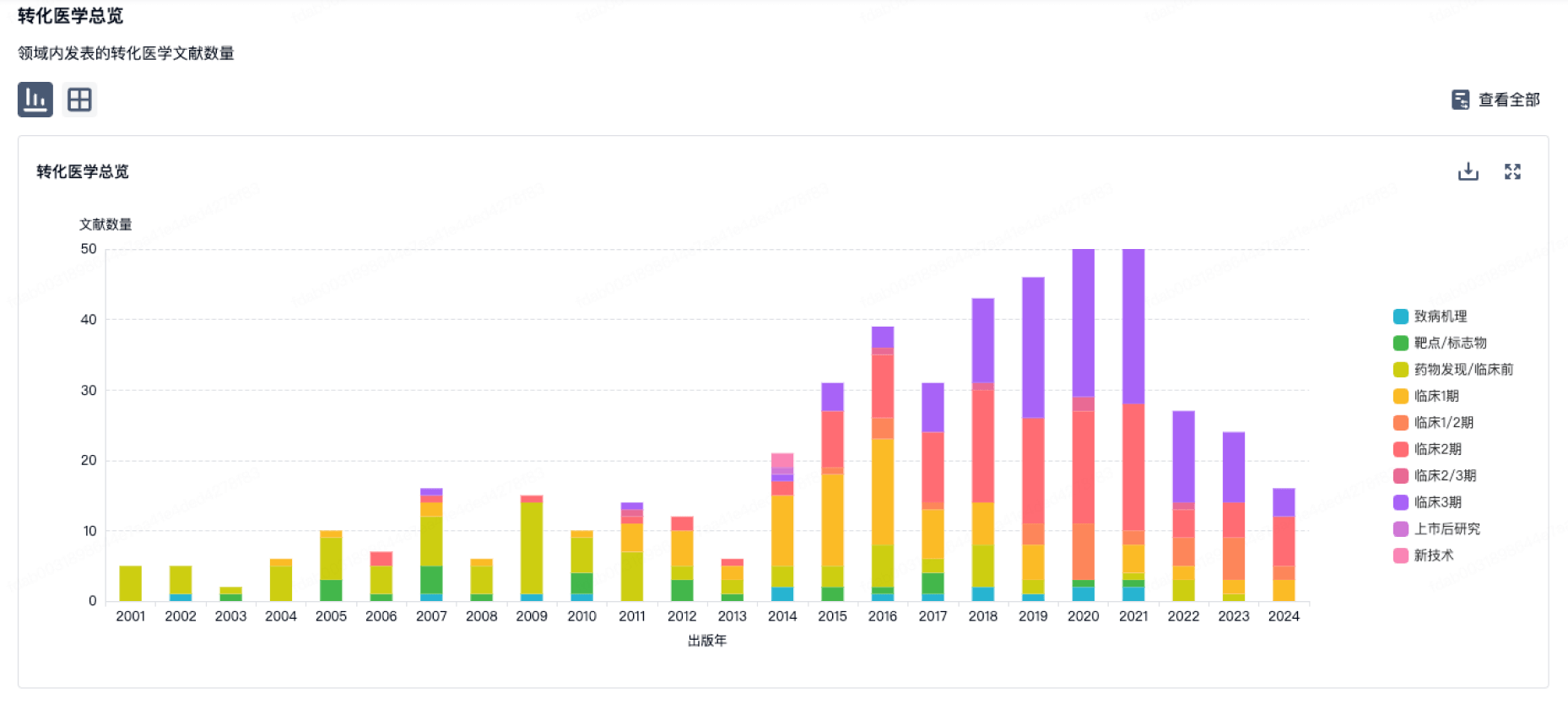
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用