预约演示
更新于:2025-05-07
CD89 x EGFR
更新于:2025-05-07
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

100 项与 CD89 x EGFR 相关的临床结果
登录后查看更多信息
100 项与 CD89 x EGFR 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2024-07-15The Journal of Immunology
Engineering Dimeric EGFR-directed IgA Antibodies Reveals a Central Role of CD147 during Neutrophil-mediated Tumor Cell Killing of Head and Neck Squamous Cancer Cells
Article
作者: Zwick, Anabel ; Braun, Felix Leon ; Linder, Manuel ; Linxweiler, Maximilian ; Weber, Lennert Jochen ; Lohse, Stefan
2024-04-01Cancer Research
Human Tumor–Associated Macrophages and Neutrophils Regulate Antitumor Antibody Efficacy through Lethal and Sublethal Trogocytosis
Article
作者: Guo, Emily ; Albelda, Steven M ; Bruns, Kyle ; Singhal, Sunil ; Bermudez, Andres ; Moon, Edmund K ; Rao, Abhishek S ; Honig-Frand, Adam ; Valerius, Thomas ; Stadanlick, Jason ; Krouse, Ryan ; Georgiou, George ; Eruslanov, Evgeniy B ; Arambepola, Sachinthani ; Sullivan, Neil T
2002-02-01Journal of Urology1区 · 医学
EPIDERMAL GROWTH FACTOR RECEPTOR AND G250: USEFUL TARGET ANTIGENS FOR ANTIBODY MEDIATED CELLULAR CYTOTOXICITY AGAINST RENAL CELL CARCINOMA?
1区 · 医学
Article
作者: ELS¨ASSER, D. ; KÜHN, R. ; van de WINKEL, J.G.J. ; KALDEN, J.R. ; STADICK, H. ; SCHROTT, K.M. ; GLENNIE, M.J. ; VALERIUS, T. ; STOCKMEYER, B. ; GRAMATZKI, M.
分析
对领域进行一次全面的分析。
登录
或
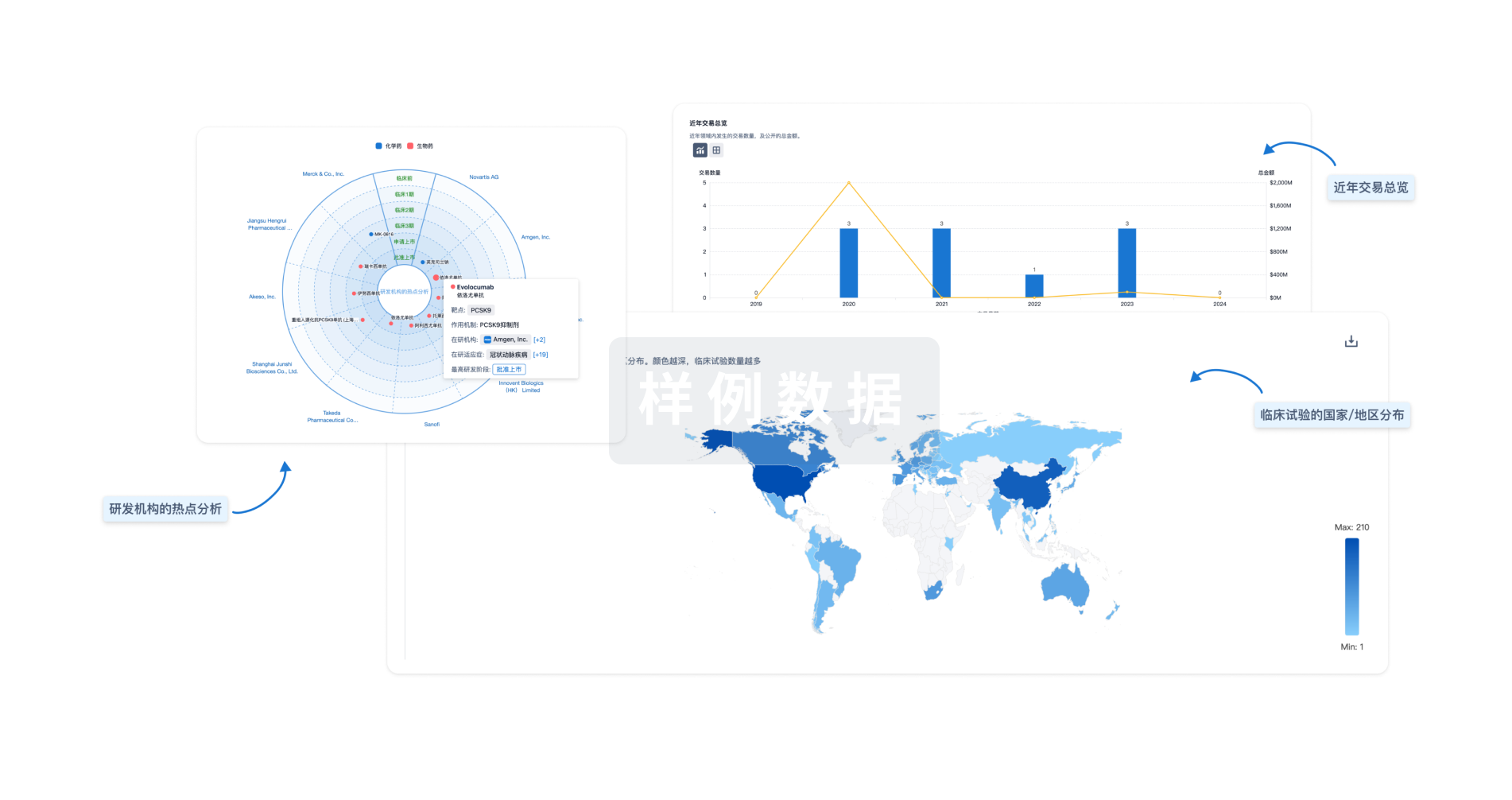
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用