预约演示
更新于:2026-07-25
Nucresiran
更新于:2026-07-25
概要
基本信息
药物类型 siRNA |
别名 ALN TTRSC04、ALN-TTRSC04 |
靶点 |
作用方式 抑制剂 |
作用机制 TTR抑制剂(转甲状腺素蛋白抑制剂)、RNAi(RNA干扰) |
非在研适应症- |
非在研机构- |
权益机构- |
最高研发阶段临床3期 |
首次获批日期- |
最高研发阶段(中国)临床3期 |
特殊审评孤儿药 (美国) |

登录后查看时间轴
结构/序列
使用我们的RNA技术数据为新药研发加速。
登录
或
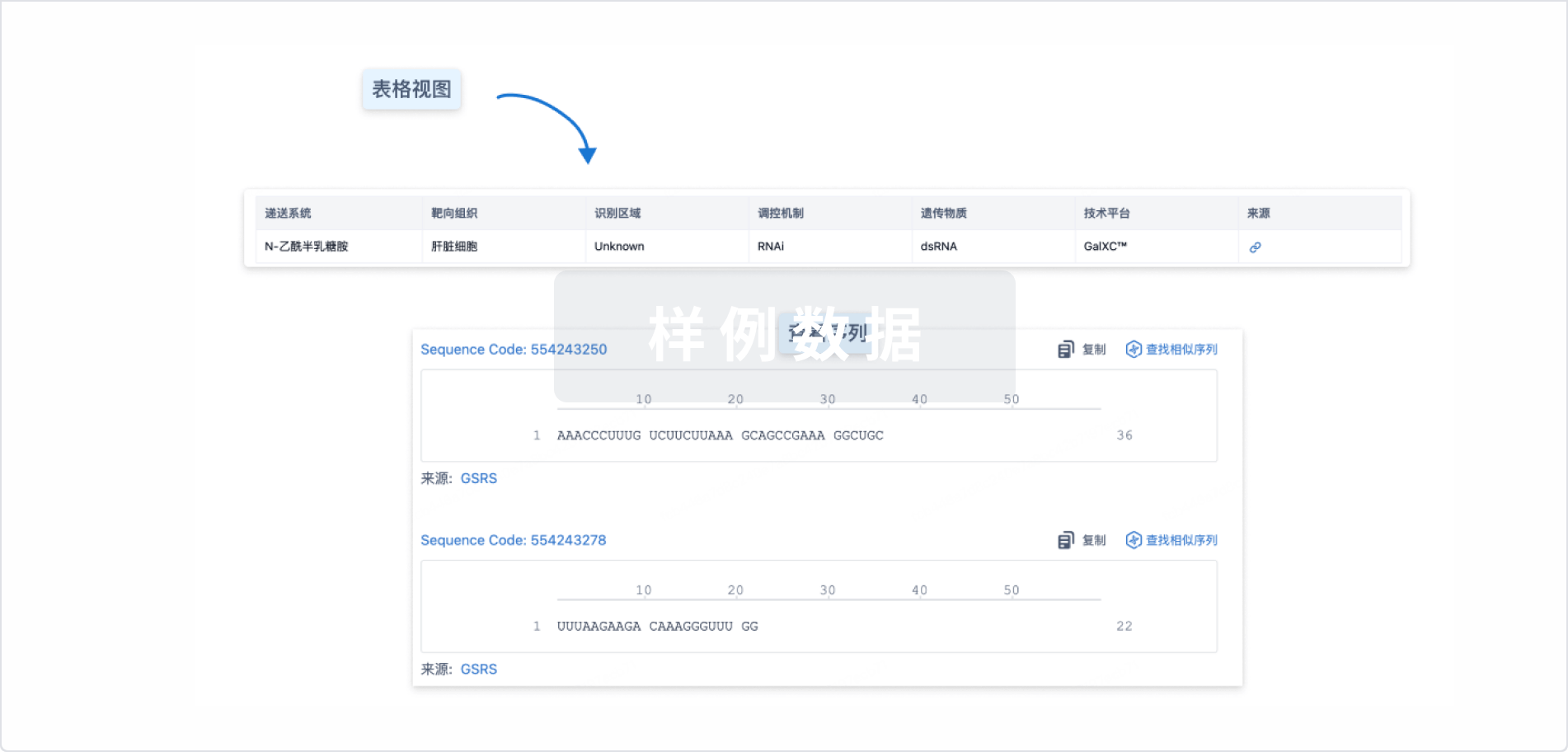
Sequence Code 227508574

来源: *****
Sequence Code 237409699
来源: *****
关联
3
项与 Nucresiran 相关的临床试验NCT07223203
TRITON-PN: A Phase 3, Global, Randomized, Open-Label Study to Evaluate the Efficacy and Safety of Nucresiran in Patients With Hereditary Transthyretin-Mediated Amyloidosis With Polyneuropathy (hATTR-PN)
The purpose of this study is to:
* Determine the efficacy of nucresiran in patients with hATTR-PN by evaluating the effect on neurologic impairment, quality of life, nutritional status, disability, and gait speed
* Demonstrate superiority of nucresiran compared to in-study vutrisiran with respect to serum transthyretin (TTR) levels
* Determine the efficacy of nucresiran in patients with hATTR-PN by evaluating the effect on neurologic impairment, quality of life, nutritional status, disability, and gait speed
* Demonstrate superiority of nucresiran compared to in-study vutrisiran with respect to serum transthyretin (TTR) levels
开始日期2025-12-12 |
NCT07052903
TRITON-CM: A Phase 3 Global, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Nucresiran in Patients With Transthyretin-Mediated Amyloidosis With Cardiomyopathy (ATTR Amyloidosis With Cardiomyopathy)
The purpose of this study is to:
* Evaluate the efficacy of nucresiran compared to placebo on reducing all-cause mortality and cardiovascular (CV) events
* Evaluate the efficacy of nucresiran compared to placebo on additional assessments of CV events and/or death
* Evaluate the efficacy of nucresiran compared to placebo on patient-reported health status and health-related quality of life
* Evaluate the efficacy of nucresiran compared to placebo on reducing all-cause mortality and cardiovascular (CV) events
* Evaluate the efficacy of nucresiran compared to placebo on additional assessments of CV events and/or death
* Evaluate the efficacy of nucresiran compared to placebo on patient-reported health status and health-related quality of life
开始日期2025-07-02 |
NCT05661916
A Phase 1, Randomized, Double-Blind, Placebo Controlled, Single-Ascending Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics (PK), and Pharmacodynamics (PD) of Subcutaneously Administered ALN-TTRSC04 in Healthy Subjects
The purpose of this study is to evaluate the safety, tolerability, pharmacodynamics (PD) and pharmacokinetics (PK) of single ascending doses of ALN-TTRSC04.
开始日期2023-01-16 |
100 项与 Nucresiran 相关的临床结果
登录后查看更多信息
100 项与 Nucresiran 相关的转化医学
登录后查看更多信息
100 项与 Nucresiran 相关的专利(医药)
登录后查看更多信息
105
项与 Nucresiran 相关的新闻(医药)2026-07-10
iStock,
Matthew Malczewski
The failure of AstraZeneca and Ionis’ Wainua in a late-stage study of ATTR-CM casts doubt on Alnylam’s next-generation candidate but is good news for others in the space, including BridgeBio and Intellia Therapeutics.
The surprising late-stage failure of AstraZeneca and Ionis’ antisense oligonucleotide Wainua in transthyretin amyloidosis cardiomyopathy this week has shaken the field, sowing doubt in certain therapies while building investor confidence in others.
“The study here was expected to work since we know TTR [transthyretin] silencers are effective,” Stifel told investors in a Thursday note, referring to Wainua’s mechanism of suppressing the expression of the TTR protein, which is misfolded in ATTR-CM, leading to pathologic clumps in the heart.
“This is absolutely the most complicated possible outcome” for Alnylam, Stifel analysts wrote, referring to the biotech’s FDA-approved Amvuttra that’s indicated for both hereditary ATTR-CM and polyneuropathy in transthyretin amyloidosis (ATTR). Alnylam is also advancing its RNAi candidate nucresiran for these conditions. Both programs work similarly to Wainua by silencing TTR.
AstraZeneca and Ionis’ setback on Thursday “represents a near-term ‘win’ for ALNY by leaving Amvuttra as the only approved TTR silencer for ATTR-CM,” Truist Securities explained in a Thursday note. But the failure “raises questions about outlook for ALNY’s next-generation silencer, nucresiran, and its ability to demonstrate incremental clinical benefit in a treatment landscape increasingly dominated by background stabilizer therapy.”
Earnings
‘Very Impressive’ Amvuttra ATTR-CM Sales Send Alnylam Soaring
In its first commercial quarter for ATTR-cardiomyopathy, Alnylam’s Amvuttra reached roughly 1,400 patients and made more than $490 million.
August 1, 2025
·
2 min read
·
Tristan Manalac
Read more
Alnylam is studying nucresiran in the
Phase 3 TRITON-CM study
, which is currently recruiting patients and expected to readout in 2030. The biotech “remains very confident” in nucresiran and TRITON, Oppenheimer wrote Thursday after a Q&A session with the company.
In particular, the firm pointed to three key advantages emphasized by Alnylam: nucresiran is a better molecule than Wainua in potency, durability and onset; TRITON-CM is a larger trial than AstraZeneca and Ionis’ study; and Alnylam has a leadership team with proven experience. “We remain positive on ALNY ahead of TRITON,” Oppenheimer said.
Alnylam ended Thursday’s trading session down 3.3%, while BridgeBio, on the other hand, swelled 15%.
Tailwind for some
AstraZeneca and Ionis’ failure “simplifies the ATTR-CM landscape,” Mizuho Securities wrote in a Thursday note focused on BridgeBio. The biotech owns the TTR stabilizer Attruby, approved in
November 2024
for ATTR-CM. Unlike Wainua and Amvuttra, which reduce the expression of TTR,
Attruby
instead binds to the protein and prevents a crucial step in the formation of the disease-causing clumps.
Earlier this month, BridgeBio
presented a post-hoc analysis
of Phase 2 and Phase 3 data for Attruby, pointing to the drug’s potential to preserve kidney function in ATTR-CM—a key renal benefit that could set it apart from other therapies in this space.
Another drug that works similarly to Attruby is Pfizer’s Vyndaqel/Vyndamax, which was
approved in 2019
and made
$1.7 billion worldwide
last year. But Phase 3 data in May suggest that Attruby has a
survival edge
in ATTR-CM.
Cardiovascular disease
BridgeBio stands up to Pfizer in ATTR-CM as late-stage data hint at better survival
Indirect comparisons between BridgeBio’s Attruby and Pfizer’s tafamidis products showed a numerical survival benefit with the biotech’s drug.
May 12, 2026
·
2 min read
·
Tristan Manalac
Read more
There has been no direct head-to-head comparison between the two medicines, but Mizuho nevertheless noted that Attruby is a better option in the hospital. “Because ATTR-CM is still a fatal disease, physicians should opt for the more potent stabilizer, BBIO’s Attruby.”
“Peak sales estimates for Attruby may edge higher” with Wainua out of the picture, the group added. The drug brought in
$362.4 million
in 2025.
Also in the ATTR-CM arena is Intellia Therapeutics, a biotech advancing the CRISPR-based gene therapy nexiguran ziclumeran (nex-z). Two Phase 3 studies of nex-z
were subject to an FDA hold
last October after a patient in one of the trials developed life-threatening liver enzyme elevations. The agency has since
fully lifted
the pause.
Nex-z works by deactivating the
TTR
gene and reducing the overall expression of the protein—much like Wainua and other silencer therapies. What sets Intellia’s candidate apart, however, is that it’s designed to be a one-time therapy. The biotech is advancing nex-z
in collaboration
with Regeneron.
The Wainua fail delivers an incremental positive to nex-z, Jefferies wrote in a Thursday note, reinforcing the analysts’ view “that achieving deep+consistent TTR knockdown is needed to drive benefit on outcomes.”
“Beyond stabilizer progression, we still expect ATTR [patients] to look for the next best option,” the analysts said.
临床3期上市批准临床结果基因疗法
2026-07-09
iStock,
Nuthawut Somsuk
The late-stage miss is “surprising,” Stifel analysts said, given that Wainua’s mechanism of silencing transthyretin protein expression has previously proven effective.
AstraZeneca and Ionis Pharmaceuticals’ antisense therapeutic Wainua failed to show significant cardiovascular benefit in a Phase 3 study of patients with transthyretin-mediated amyloid cardiomyopathy.
The companies didn’t provide specific data in their
news release Thursday morning
, revealing only that Wainua, when added to standard of care, “did not meet the primary efficacy endpoint,” which was a composite of cardiovascular (CV) mortality and recurrent CV clinical events vs. placebo after up to 140 weeks of follow-up.
The failure is “surprising,” Stifel analysts told investors in a Thursday note. “The study here was expected to work since we know TTR [transthyretin] silencers are effective,” the firm added. H.C. Wainwright, in a June 25 note, ascribed a “75% probability of approval” for Wainua in transthyretin-mediated amyloid cardiomyopathy (ATTR-CM).
Wainua in ATTR-CM was “a key valuation driver” for Ionis, Stifel noted Thursday, adding that the disappointing outcome is “obviously a negative” for the biotech. Ionis is down 20% in premarket U.S. trading, while AstraZeneca took a 9% hit on the British stock exchange.
The partners will conduct a full analysis of the results and present their findings at an upcoming medical congress next month.
Wainua was
first approved in December 2023
for the treatment of polyneuropathy associated with hereditary transthyretin-mediated amyloidosis (hATTR-PN). The drug was originally discovered and developed by Ionis and came to AstraZeneca in December 2021 when the pharma dropped
$200 million upfront
for the right to jointly advance and commercialize the product. This deal also included a $485 million payment upon regulatory approvals and up to $2.9 billion in sales-related milestones, plus royalties.
The companies ran
CARDIO-TTRansform
to assess the therapeutic value of Wainua in patients with ATTR-CM. The trial enrolled more than 1,400 patients who were already receiving available standard of care, and who were then randomly assigned to receive add-on Wainua or placebo.
AstraZeneca and Ionis on Thursday made no mention of their regulatory strategy going forward for Wainua in ATTR-CM. However, Stifel in its note said that “trying to approach regulators here on these data would seem like a stretch.”
CARDIO-TTRansform’s disappointing result comes after
BridgeBio
last week released posthoc data suggesting that its transthyretin stabilizer Attruby preserved kidney function in patients with ATTR-CM—a benefit that analysts say could set it apart from other therapies in this space. BridgeBio is up nearly 10% in pre-market trading Thursday.
The kidney benefit “adds differentiation” to Attruby, Jefferies said in a July 2 note, adding that the magnitude of renal improvements even “looks competitive to kidney-targeted therapies.
“Cardiorenal protective effects seem distinct from other therapies,” Jefferies added, pointing to Pfizer’s
Vyndaqel/Vyndamax
. “Ultimately, slowing down kidney progression could improve hospitalization/mortality outcomes.”
Rare diseases
BridgeBio touts kidney benefits for Attruby, differentiating from amyloidosis competitors
BridgeBio’s Attruby preserves kidney function in patients with transthyretin amyloidosis cardiomyopathy, an effect that is “distinct” from other drugs in this space, according to Jefferies.
July 6, 2026
·
2 min read
·
Tristan Manalac
Read more
Meanwhile, the failure of Ionis and AstraZeneca’s trial is “absolutely the most complicated possible outcome” for another key competitor, Alnylam, which markets Amvuttra for ATTR-CM, Stifel wrote.
On the one hand, “not having [Wainua] launch in TTR-CM in 2027 should represent a significant tailwind for vutrisiran relative to expectations,” the analysts wrote. However, Alnylam has another asset, nucresiran, in Phase 3 trials for the disease. The general expectation is that patients in Alnylam’s trial will be on background therapy, as was the case for AstraZeneca and Ionis’ trial, which Stifel said it had been watching for “to affirm this combo synergy.”
Alnylam shares were up nearly 18% in premarket trading on Thursday.
上市批准临床3期AHA会议
2026-06-20
·孙泽华
阿尔尼拉姆投资深度分析
(注意本文大量使用AI资料,本人初步复核,部分数据可能未做复核,请各位谨慎看待)
1. 公司概况
Alnylam Pharmaceuticals(纳斯达克:ALNY)是全球RNA干扰(RNAi)疗法的开创者与领导者,创立于2002年,总部位于美国马萨诸塞州剑桥市。公司由诺贝尔奖得主Andrew Fire和Craig Mello的RNAi基础科学成果发展而来,历经二十余年的持续投入,已成功将RNAi技术从学术概念转化为获批上市的商业药物,并于2025年首次实现全面盈利,标志着公司进入商业化成熟阶段。
Alnylam的战略目标是通过RNAi技术"让不可成药的靶点变得可成药",专注于具有高度未满足需求的遗传性疾病、心血管疾病和神经系统疾病。公司目前拥有5个已获批上市产品(另有Leqvio由诺华商业化,合计6个上市RNAi产品)、7个三期临床项目,并建立了全球最大的RNAi研发管线。
公司发展历程关键里程碑
年份
关键事件
战略意义
2002
公司创立,由诺贝尔奖得主Andrew Fire和Craig Mello的RNAi基础科学成果发展而来,Phillip Sharp等MIT/哈佛科学家联合创办
RNAi疗法领域奠基,将诺贝尔级科学发现转化为产业
2004
纳斯达克IPO上市(ALNY);首批GalNAc-siRNA临床前数据发表
资本市场验证+核心递送技术突破
2005
与罗氏(Roche)签署$3.3亿合作,获RNAi全球许可
首次大型药企合作,验证RNAi平台价值
2010
与赛诺菲签署$7亿+战略合作(罕见病RNAi领域)
扩大合作伙伴矩阵,获取关键研发资金
2013
Revusiran(一代TTR RNAi)进入三期临床
首次挑战大规模适应症,但最终失败
2016
Revusiran三期失败(安全性+疗效不足),股价暴跌~50%
公司至暗时刻,但也促使ESC+化学平台和GalNAc递送技术的全面升级
2017
Patisiran(ONPATTRO)APOLLO三期达到主要终点;Givosiran二期数据积极
从Revusiran阴影中走出,RNAi概念再次验证
2018
Onpattro(patisiran)获FDA批准,全球首个siRNA药物;获FDA突破性疗法认定
历史性里程碑——RNAi从科学概念变为成药
2019
Givlaari(givosiran)获FDA批准;诺华以$97亿收购TMC(The Medicines Company)从而获得Inclisiran(Leqvio)所有权;Oxlumo三期成功
商业模式验证(收购vs授权+自主推进并行),管线深度证明
2020
Oxlumo(lumasiran)获FDA批准;与黑石(Blackstone)签署$20亿战略融资
罕见病产品矩阵拓展;战略融资增强财务实力
2021
Vutrisiran(AMVUTTRA)HELIOS-A三期达到终点;与罗氏签署zilebesiran合作(美国50/50利润分享)
二代TTR产品蓄势待发,高血压大适应症布局
2022
Amvuttra(vutrisiran)获FDA批准用于hATTR-PN;Leqvio(inclisiran)获FDA批准用于HeFH/ASCVD
第二代TTR产品上市,LNP→GalNAc皮下注射迭代成功
2023
HELIOS-B(AMVUTTRA ATTR-CM适应症)三期达到主要终点:全因死亡率降低36%
打开ATTR-CM$100亿级市场大门,数据优于辉瑞Tafamidis
2024
Nucresiran(ALN-TTRsc04)一期数据优异:TTR降低>90%,半年一次给药;BridgeBio Acoramidis获FDA批准ATTR-CM
下一代TTR产品蓄力;ATTR赛道竞争加剧
2025年3月
Amvuttra获FDA批准扩展ATTR-CM适应症;Qfitlia(fitusiran)获FDA批准血友病A/B
最大商业化机遇开启,第6个获批产品
2025全年
首次实现GAAP/Non-GAAP双重盈利,年收入$37亿;Nucresiran启动TRITON三期;Zilebesiran推进ZENITH三期
公司转型为成熟盈利企业,三大新增长极(TTR下一代+高血压+补体)布局完成
2. 公司股权结构
公司持股高度机构化,股东以投资机构持股为主。高管持股比例偏低(加起来可能不到1%),持股仅为10–20万股,市值不到1000万美元,相当于一年薪酬水平。
股东名称
持股数量(估)
持股比例
持仓市值FMR LLC(富达)
1,458万股
~11.0%
~$69亿Vanguard Group
1,355万股
~10.3%
~$64亿Capital World Investors
1,080万股
~8.3%
~$51亿Capital Research Global Investors
~540万股
~4.1%
~$26亿BlackRock
~500万股
~3.8%
~$24亿Geode Capital Management
~300万股
~2.3%
~$14亿其他机构
—
~55.3%
—内部人持股(管理层+董事)
~10–20万股
<1%
<$1,000万公众自由流通
—
~3.5%
—
3. 核心高管团队
姓名
职位
背景亮点Yvonne Greenstreet
, MD, MBA, OBE
首席执行官(CEO)
前AstraZeneca、Pfizer高管;英国大英帝国勋章获得者;2022年接任CEO,主导公司商业化转型,带领公司实现首次盈利Kevin Fitzgerald
, PhD
首席科学官(CSO)兼EVP,研究负责人
Alnylam联合创始人之一;RNAi递送领域顶级科学家;主导ESC+化学修饰平台和GalNAc递送系统的开发,20余年研究领导经验Pushkal Garg
, MD
首席研发官(CRDO)
前诺华全球研发主管;负责监督Alnylam所有临床开发项目,包括关键的HELIOS-B、ZENITH等三期试验Jeff Poulton
, MBA
首席财务官(CFO)
拥有超过20年生物制药财务管理经验Tolga Tanguler
, MBA
首席商务官(CCO)
主导AMVUTTRA在ATTR-CM适应症的商业化战略,推动年收入从$9.7亿增至$23.1亿
4.SiRNA技术原理与公司技术平台4.1 siRNA药物完整作用机制详解
五步作用机制
第一步:siRNA进入细胞——合成的双链siRNA(21-23个碱基对)通过GalNAc配体介导的受体内吞进入肝细胞。三价GalNAc配体与肝细胞表面高度表达的ASGPR受体特异性结合,实现肝细胞选择性摄取,避免其他组织暴露。
第二步:RISC装载——进入细胞后,双链siRNA被Dicer酶或直接被Argonaute-2(Ago2)蛋白识别。Ago2是RNA诱导沉默复合物(RISC)的核心组分,它将siRNA的"过客链"(passenger strand)切割并丢弃,仅保留"引导链"(guide strand)。
第三步:靶mRNA识别与切割——RISC-引导链复合物在细胞质中扫描mRNA,当引导链序列与靶mRNA完全互补配对时(通常针对3'UTR或CDS区),Ago2在配对位点精确切割靶mRNA。
第四步:mRNA降解——被切割的mRNA片段被细胞内其他核酸外切酶快速降解,彻底阻止该mRNA翻译为蛋白。
第五步:RISC循环催化——关键的是,一个RISC-siRNA复合物可以反复切割多个靶mRNA分子(催化式作用),这解释了为何siRNA在极低浓度下即可实现>90%的靶蛋白沉默。这也是siRNA药物给药间隔可长达数月的分子基础。4.2 通过siRNA技术将传统"难以成药的靶点"成药
传统药物发现中,约80–90%的人类蛋白被认为"不可成药"(undruggable),主要原因包括:①缺乏小分子结合口袋——许多蛋白表面平滑,没有适合小分子结合的沟槽或口袋;②蛋白-蛋白相互作用(PPI)——许多疾病相关通路依赖于两个蛋白的相互作用,小分子难以干扰这种大面积相互作用;③细胞内靶点——抗体药物(单抗)由于分子量大,无法进入细胞内部,只能作用于细胞表面的受体或分泌蛋白;④缺乏催化活性位点——许多疾病相关蛋白是"假酶"或调节蛋白,没有可被小分子抑制的活性位点。
siRNA技术通过直接靶向mRNA,旁路了上述所有限制:只要靶蛋白有mRNA编码,就可以设计siRNA序列与之结合并降解,无需蛋白有"结合口袋"或"活性位点";siRNA通过GalNAc配体介导的细胞内吞进入细胞,可直接作用于细胞内靶点;且siRNA的"序列特异性"使其可以区分高度同源的蛋白家族成员,实现传统小分子无法达到的选择性。4.3 公司技术平台
公司整套技术分为siRNA化学修饰平台、靶向递送平台、多功能分子工程平台、生产制造平台四大板块,核心壁垒是三代GalNAc肝靶向递送,同时布局LNP、C16中枢、IKARIA外周组织、GEMINI双靶点等差异化管线平台,支撑5款上市RNAi药物与全器官管线开发。
平台
技术内容
代际进展
代表产品siRNA化学修饰平台
对分子结构进行定向改造,提升siRNA稳定性,减少免疫反应与脱靶效应,延长药效周期
STC → ESC → ESC+(第三代),长效给药,皮下注射可行
AMVUTTRA、GIVLAARI、OXLUMO(均为ESC+)靶向递送平台(肝脏)
GalNAc三价偶联:靶向性强、皮下给药、安全性高,为主流商业化路线;LNP脂质纳米颗粒:早期路线,需静脉输注
LNP(一代)→ GalNAc(二代主流)
GalNAc:AMVUTTRA等;LNP:ONPATTRO(逐步淘汰)靶向递送平台(肝外)
C16脂质偶联(中枢神经)、IKARIA平台(外周组织)——攻克跨屏障、多器官靶向难题
临床前及早期临床阶段
神经系统、肌肉、心脏等在研管线GEMINI双靶点siRNA平台
单分子同时沉默两个致病基因,适配复杂疾病治疗
临床前研发阶段
复杂代谢与心血管疾病siRELIS酶促合成平台
自研生产工艺,降低成本、提升产能,为药物规模化量产提供保障
商业化规模生产
支撑全线已上市产品
与CRISPR的本质区别
RNAi作用于mRNA水平(转录后调控),不改变DNA序列,效应可逆;CRISPR直接编辑基因组DNA,效应永久不可逆。相比小分子药物(仅能针对约10–20%的蛋白靶点)和抗体药物(仅能作用于细胞外靶点),siRNA理论上可靶向几乎所有有mRNA编码的蛋白,"可成药靶标空间"扩大10倍以上。
若Intellia NTLA-2001在ATTR领域获批,其"一次性治疗"标签将分流部分患者。但CRISPR的永久性编辑在ATTR(老年慢病)领域的安全性接受度仍有待验证,预计2030年前RNAi仍将是ATTR市场的主导疗法。技术平台竞争优势
Alnylam RNAi技术平台 vs 竞争技术路线比较
5.重点产品与在研管线5.1 已上市产品
AMVUTTRA(Vutrisiran)—— 旗舰产品,全球ATTR市场核心驱动力
ATTR-CM适应症是公司巨大增长机会。 ATTR心肌病全球患者估计超过30万人(重度漏诊),而此前仅有辉瑞tafamidis(Vyndaqel/Vyndamax)获批。2025年3月,AMVUTTRA基于HELIOS-B三期临床数据(死亡率降低36%,首次心血管事件降低38%)获FDA批准,成为唯一同时获批PN和CM适应症的ATTR治疗药物。Q4 2025单季收入即已达$8.27亿,年化接近$33亿,仍处快速上升通道。5.2 重点在研管线ALNY在研管线中心从罕见病转向高血压、MASH、糖尿病等大适应症。靶点也拓展到肝以外的靶点。
Mivelsiran 两个适应症、ALN-HTT02、ALN-5288、ALN-SOD3、ALN-SNCA。
6.各产品与管线竞争格局分析6.1 ATTR淀粉样变性市场竞争格局6.1.1 ATTR疾病背景
转甲状腺素蛋白(TTR)淀粉样变性概述
疾病机制:转甲状腺素蛋白(Transthyretin, TTR)是由肝脏合成的转运蛋白(运输甲状腺素和视黄醇)。正常TTR为四聚体结构,当四聚体解离为单体后,单体错误折叠并聚合形成淀粉样纤维,沉积在器官组织中导致器官功能损伤。根据突变状态可分为:遗传型(hATTR/ATTRv)——由TTR基因突变引起(>130种已知突变),发病年龄30-70岁;野生型(ATTRwt/wtATTR)——无基因突变,随年龄增长自然发生四聚体不稳定,主要影响65岁以上老年人。ATTR-CM(心肌病型)病理
:TTR淀粉样纤维沉积于心肌间质,导致心室壁增厚、舒张功能障碍、进行性心力衰竭流行病学
:全球确诊患者约30-50万,但漏诊率极高(估计5-10倍),美国确诊约12万,以65岁以上男性为主(wtATTR占ATTR-CM约80%)预后
:未经治疗的中位生存期2-5年(wtATTR),4-6年(hATTR-CM);主要死亡原因为心衰和猝死诊断
:核素扫描(99mTc-PYP)为金标准无创诊断;心脏MRI+血/尿免疫固定电泳排除AL型;确诊率正在快速提升市场
:辉瑞Tafamidis 2025年销售额$63.8亿,为ATTR-CM市场主导;全球ATTR-CM药物市场预计2030年突破$120亿附:辉瑞Tafamidis(Vyndaqel/Vyndamax)过去10年全球销售收入
年份
全球销售额
(亿美元)
同比增长
备注
2015
~$2–3亿
—
欧盟市场为主,ATTR-CM适应症尚未获批
2016
~$3–4亿
~+20%
欧洲市场稳步增长
2017
~$4–5亿
~+25%
逐步渗透确诊患者
2018
~$5–6亿
~+20%
美国上市前夜,欧洲市场持续增长
2019
~$8–10亿
~+60%
美国FDA批准ATTR-CM适应症,市场快速放量
2020
~$15亿
~+50%
美国市场全面铺开
2021
~$22亿
~+47%
持续渗透,诊断为ATTR-CM患者增加
2022
~$28亿
~+27%
市场主导地位巩固
2023
~$33亿
~+18%
增速放缓但持续成长
2024
$54.5亿
+65%
Vyndamax(每日一次)替代Vyndaqel,依从性提升推动增长
2025
$63.8亿
+17%
预计2025全年,辉瑞财报指引区间中值
数据来源:辉瑞年报、医药行业分析报告、SEC财报文件。Tafamidis(Vyndaqel/Vyndamax)自2019年美国获批ATTR-CM适应症后,销售收入持续大幅增长,确诊率提升+治疗渗透率提升双轮驱动。ATTR-PN(多发性神经病变型)病理
:TTR淀粉样纤维沉积于周围神经和自主神经系统,导致进行性感觉运动神经病变和自主神经功能障碍流行病学
:全球hATTR-PN确诊患者约1-1.5万人,为超罕见病;葡萄牙、瑞典、日本为高发区(创始人效应)预后
:未经治疗的进行性恶化,10年内丧失行走能力;自主神经病变(体位性低血压、腹泻、性功能障碍)严重影响生活质量诊断
:组织活检(脂肪垫/神经)+ TTR基因检测;较CM更容易漏诊市场
:市场规模约$25-30亿,AMVUTTRA已占据主导地位,ONPATTRO被逐步替代6.1.2 ATTR-CM核心竞争产品疗效与安全性对比
ATTR-CM是核心战场:年销售额有望突破$100亿
全球ATTR-CM患者估计超过30万人(主要在65岁以上老年人群中),且严重漏诊,真实患者数可能是确诊数的5–10倍。随着诊断率提升和AMVUTTRA快速放量,这一市场有望成为生物科技史上最大罕见病市场之一。
产品
机制
给药
关键临床数据(疗效)
安全性特征AMVUTTRA(Vutrisiran)
RNAi,TTR mRNA降解
每季度皮下注射
HELIOS-B:全因死亡率↓36%(42个月随访);CV事件↓38%;6MWD改善;KCCQ-OS改善;唯一"根源疗法"——从根本上阻断TTR蛋白生成
注射部位反应(~10%);肝酶升高少见;无致畸性证据;可逆(停药后TTR恢复)Vyndamax(Tafamidis)
辉瑞
TTR四聚体稳定剂(小分子)
每日1粒口服
ATTR-ACT:全因死亡率↓30%(30个月);CV住院↓32%;不阻断TTR生成,仅减缓四聚体解离——"对症稳定"而非"根源治疗"
总体耐受性好;腹泻(~11%);局限性:对已形成淀粉样沉积无清除作用;疗效受TTR突变类型影响(V122I突变响应较弱)Attruby(Acoramidis)
BridgeBio
TTR四聚体稳定剂(小分子,AG10)
每日两次口服
ATTRibute-CM:全因死亡率优效(HR 0.59);CV事件复合终点优效;体外TTR稳定化效力强于Tafamidis(接近100%四聚体稳定)
总体耐受性好;局限性:每日两次vs Tafamidis每日一次,依从性劣势;作为稳定剂同样不能清除已有沉积;上市时间短,长期安全性数据有限Patisiran(ONPATTRO)
RNAi,LNP递送
每3周静脉输注
逐步被AMVUTTRA取代
输注相关反应;需皮质类固醇预给药Nucresiran(ALN-TTRsc04)
RNAi,下一代TTR
每半年皮下注射
Phase 1:TTR降低>90%;TRITON三期进行中
安全性数据尚在收集中NTLA-2001(Nexiguran)
Intellia
CRISPR/Cas9体内基因编辑
静脉输注(LNP)
Phase 1:TTR降低>90%;理论上一次治疗永久沉默TTR脱靶编辑风险、永久不可逆、LNP肝毒性、免疫原性——慢性病长期安全性存疑
6.1.3 AMVUTTRA vs Tafamidis vs Acoramidis:疗效与安全性深度对比✅ AMVUTTRA相对优势疗效优势
:HELIOS-B全因死亡率降低36% vs ATTR-ACT的30%(注意:跨试验比较需谨慎,但数据趋势一致优于Tafamidis)机制优势
:RNAi为"根源疗法"——从根本上阻断TTR蛋白合成(TTR降低>90%),而Tafamidis/Acoramidis仅稳定四聚体,不能阻止所有TTR解离依从性优势
:每季度1次皮下注射 vs Tafamidis每日1次口服 vs Acoramidis每日2次口服——慢性心衰老年患者多药联用,注射频率低=依从性保障广谱覆盖
:唯一同时获批PN+CM适应症的ATTR药物,医生处方无需区分亚型可逆性安全
:如需停药(手术、不良反应等),TTR蛋白水平在数周内恢复,而Tafamidis/Acoramidis停药后四聚体稳定化即刻消失⚠️ AMVUTTRA相对劣势给药方式劣势
:注射vs口服——心内科医生处方习惯偏向口服药,患者心理偏好口服价格劣势
:~$50万/年 vs Tafamidis ~$22.5万/年(WAC),2倍以上价差对医保支付构成压力安全性顾虑
:虽然可逆,但"基因沉默"概念对部分医生/患者仍存心理顾虑Acoramidis威胁
:体外数据提示其四聚体稳定化效率高于Tafamidis,若临床头对头数据证实,可能分流部分Tafamidis不响应患者Tafamidis先发壁垒
:辉瑞6年+市场教育、指南推荐、医生处方路径依赖——切换成本高肝外TTR问题
:GalNAc仅靶向肝脏,脑脊液和眼内TTR不受影响——ATTR-PN的CNS/眼部症状可能需要联合治疗6.1.4 Acoramidis vs Tafamidis:二代稳定剂竞争
Acoramidis(AG10)——Tafamidis的"升级版"稳定剂
Acoramidis优势:①体外TTR稳定化效率接近100%(Tafamidis约70-80%),理论上可实现更完全的四聚体稳定;②ATTRibute-CM试验显示全因死亡率HR 0.59,数值上优于ATTR-ACT的HR 0.70(但跨试验比较需谨慎);③对V122I突变(非洲裔常见,对Tafamidis响应较弱)可能更有效。
Acoramidis劣势:①每日两次口服 vs Tafamidis每日一次——依从性劣势明显,尤其对老年多药联用患者;②上市时间晚5年(2024 vs 2019),Tafamidis已建立强大处方惯性;③长期安全性数据仅有1-2年 vs Tafamidis 6年+;④BridgeBio商业化能力远不如辉瑞,可能通过授权给大药企分销。
对AMVUTTRA的影响:Acoramidis主要侵蚀Tafamidis份额(同属口服稳定剂类),对AMVUTTRA的注射剂型差异化影响有限。但Acoramidis若成功证明"口服也可达到接近RNAi的疗效",可能推迟部分患者从口服切换到注射的意愿。
竞争优势小结:AMVUTTRA是唯一同时拥有PN+CM适应症的ATTR药物,且HELIOS-B数据在死亡率改善上显示出全因死亡率降低36%的优越疗效。相比每日口服的TTR稳定剂,AMVUTTRA的季度一次皮下注射在依从性上具有显著优势,且作为"根源疗法"(从根本上阻断TTR生成)相比稳定剂的理论优越性已获临床数据证实。然而,Tafamidis的先发优势+口服便利+价格优势,以及Acoramidis的"更强稳定剂"定位,仍将在2026-2030年间构成实质性竞争。预计AMVUTTRA在ATTR-CM治疗格局中将从"新进入者"逐步过渡到"联合治疗首选",与Tafamidis/Acoramidis形成"RNAi+稳定剂"双药联用趋势。6.2 高血压市场(Zilebesiran)竞争详细分析6.2.1 高血压疾病背景与治疗现状
高血压:全球最大慢性病之一,未控患者仍超10亿
全球约13-14亿成年人患有高血压(≥140/90 mmHg),其中中国约3亿、美国约1.2亿患者。高血压是中风、心肌梗死、心衰、慢性肾病的最主要可预防危险因素。尽管一线降压药广泛应用,全球仍有约50-60%患者血压未达标(<130/80 mmHg),其中约10-15%(全球1.3-2亿)属于"难治性高血压"(≥3种降压药联用仍不达标),即Zilebesiran的核心目标人群。6.2.2 现有高血压药物类型、机制与代表产品6.2.3 RAAS系统原理与现有药物无法满足的需求
RAAS系统:Zilebesiran的革命性靶点
RAAS系统全称:肾素-血管紧张素-醛固酮系统(Renin-Angiotensin-Aldosterone System)。
完整通路:①肾脏感知低血压→释放肾素(Renin);②肾素切割肝脏分泌的血管紧张素原(AGT)→生成血管紧张素I(Ang I);③血管紧张素转化酶(ACE)将Ang I→血管紧张素II(Ang II,最强效血管收缩肽);④Ang II结合AT1受体→血管收缩+醛固酮分泌(水钠潴留)→血压升高;⑤醛固酮过量还会促进心脏重构和肾脏损伤。
现有RAS药物阻断位置:①ACEI:阻断Ang I→Ang II(位置③);②ARB:阻断Ang II→AT1受体(位置④);③肾素抑制剂:阻断肾素→Ang I(位置②,但已被证明安全性不足)。这三种药物均"不完全阻断"——如果上游过度激活(高肾素血症),下游Ang II生成仍可绕过药物阻断。
Zilebesiran的革命性:直接靶向AGT(位置②的上游源头)——通过siRNA沉默肝脏AGT合成,使血浆AGT降低>90%。由于AGT是RAS系统的"底物源头",理论上可以比ACEI/ARB/肾素抑制剂更完全地阻断RAS过度激活。且每6个月注射一次的依从性优势,对全球50%以上血压未达标患者具有极强的临床价值。6.2.4 高血压用药顺序与市场规模标准用药顺序(指南推荐)
一线:ACEI/ARB + CCB/利尿剂(双药联用),单药首选 ACEI 或 ARB
二线:ACEI/ARB + CCB + 利尿剂(三药联用)
难治性高血压:三药联用 + 螺内酯(MRA,第4种药)
血压仍未达标:考虑第5种药,或排除继发性高血压未满足需求
:约10-15%患者即便使用4-5种药仍不达标;依从性差(每日口服,50%+患者1年内自行减药/停药)市场规模与Zilebesiran机会
全球降压药市场:~$250-260亿/年(2024E)
其中通用药占70-80%(价格极低)
Zilebesiran目标:难治性/未控高血压(全球~1,300-2,000万人)
定价假设:$8,000-12,000/年(远低于AMVUTTRA但高于通用药100倍以上)
医保准入核心争议:能否证明"半年一次注射"可转化为"长期心血管事件减少",从而节约总体医疗支出6.2.5 Zilebesiran vs 现有高血压疗法竞争分析
Zilebesiran:瞄准$200亿+未控高血压蓝海市场
全球约13亿成年人患有高血压,其中仅美国就有约1.2亿患者。约10–15%的患者(全球约1,300–2,000万人)患有难治性/未控型高血压,即便使用3种以上降压药仍无法达标。Zilebesiran靶向AGT(血管紧张素原),是RAS系统最上游的靶点,半年一次皮下注射,若获批将成为高血压治疗范式的革命性突破。与罗氏共同开发(美国50/50利润分享,海外低双位数版税),ZENITH三期心血管结局试验正在推进中。
✅ Zilebesiran竞争优势
靶向AGT(RAS系统最上游),机制新颖且覆盖面广
每年仅需两次皮下注射,依从性远超每日口服(高血压患者50%+用药依从性差)
KARDIA-2/3三期数据:收缩压降低15–20mmHg,联合标准治疗效果叠加
与罗氏共同开发,拥有全球最强心血管商业化能力
REVERSIR备用解药提供独特安全保障——降压领域"过度降压"是最大安全顾虑
ZENITH心血管结局试验——若获MACE阳性,将获得指南级推荐地位⚠️ 主要挑战与竞争
高血压市场极度价格敏感:通用ACEI/ARB仅$10–30/月,Zilebesiran预计$5,000–10,000/年
需心血管结局试验(MACE终点)支撑获批,时间窗口长(2027–2028读出)
处方习惯壁垒——心内科医生对每日口服药处方路径依赖极强
医保支付方可能要求大幅折扣/风险分担方才纳入
目标人群定位(难治性/未控高血压)需要精准患者识别策略
若Intellia等CRISPR体内编辑实现"一次性治愈高血压",将构成长期颠覆性威胁6.3 补体介导疾病市场(Cemdisiran)竞争详细分析6.3.1 补体系统与C5靶点背景
补体系统:先天免疫的核心效应系统
补体系统概述:补体是血液中30余种蛋白质的级联反应系统,是先天免疫的核心组成部分。补体可通过经典途径(抗原-抗体复合物激活)、凝集素途径(病原体表面糖基识别)和替代途径(自发C3水解)三种方式激活,最终汇聚至C5转化酶将C5切割为C5a(炎症介质)和C5b(膜攻击复合物MAC组分)。过度或异常的补体激活导致自身组织损伤,是多种自身免疫和炎症性疾病的核心病理机制。
C5靶点意义:C5是补体终末通路的枢纽分子。阻断C5可同时抑制C5a介导的炎症反应和C5b-9(MAC)介导的细胞溶解。C5抑制已成为PNH、重症肌无力、非典型溶血尿毒综合征(aHUS)、视神经脊髓炎谱系疾病(NMOSD)等多种补体介导疾病的标准治疗。6.3.2 三大补体介导疾病概述PNH(阵发性睡眠性血红蛋白尿)病理
:获得性PIG-A基因突变→GPI锚定蛋白缺失→补体调节蛋白(CD55/CD59)无法锚定于红细胞膜→C5-MAC介导的血管内溶血流行病学
:全球约1.5-2万确诊患者,年发病率约1-2/100万人,超罕见病临床表现
:血管内溶血(血红蛋白尿、贫血)、血栓事件(主要死因,80%患者)、骨髓功能受损预后
:未治疗中位生存期约10年,主要死因为血栓;C5抑制剂可将血栓风险降低约80%市场规模
:2025年全球PNH治疗药物市场约$35-40亿,Soliris/Ultomiris主导gMG(全身型重症肌无力)病理
:自身抗体(抗AChR、抗MuSK)激活补体,导致神经肌肉接头突触后膜补体介导的损伤流行病学
:全球约70-100万患者,患病率约10-20/10万人临床表现
:波动性肌无力和易疲劳(眼肌、延髓、四肢),严重者呼吸肌麻痹标准治疗
:胆碱酯酶抑制剂(吡啶斯的明)+ 免疫抑制剂+ 靶向生物制剂(C5抑制剂、FcRn拮抗剂、CD20单抗)市场规模
:2025年全球gMG治疗药物市场约$40-50亿,C5抑制剂约占60%GA(地图状萎缩,晚期年龄相关性黄斑变性AMD)病理
:补体过度激活导致视网膜色素上皮细胞(RPE)损伤和萎缩,视野中心出现来进行性缺损流行病学
:全球约500-800万晚期AMD患者中有GA约100-150万(美国约100万),随人口老龄化快速增长临床表现
:中心视力进行性丧失,最终致盲;目前尚无治愈手段,仅可延缓进展治疗进展
:2023年Apellis Pegcetacoplan(C3抑制剂)和Iveric Bio/Astellas Avacincaptad(C5抑制剂)获批,为首批GA治疗药市场规模
:2025年全球GA治疗药物市场约$5-10亿,预计2030年达$50-80亿(大量未满足需求)6.3.3 现有补体药物类型、机制与代表产品6.3.4 补体疾病用药顺序与未满足需求PNH标准治疗路径
一线:C5单抗(Soliris→Ultomiris),血栓风险降低80%,溶血完全控制
C5抑制剂无效/不耐受:切换B因子抑制剂(Iptacopan)或C3抑制剂未满足需求
:①静脉输注频率(即便Ultomiris每8周仍需医院输注);②Extravascular溶血(C5抑制无法完全阻止);③感染风险(脑膜炎奈瑟菌)gMG标准治疗路径
一线:胆碱酯酶抑制剂+糖皮质激素+免疫抑制剂
二线:靶向生物制剂(抗CD20、新生儿FcRn拮抗剂、C5抑制剂)未满足需求
:①约30-40%患者对现有生物制剂响应不足;②静脉输注负担重;③长期免疫抑制感染风险6.3.5 Cemdisiran竞争详细分析
Cemdisiran:RNAi进军补体疾病——C5蛋白皮下敲降的差异化路径
Cemdisiran是靶向补体C5的siRNA疗法,每月一次皮下注射,可抑制肝脏中C5蛋白的合成。与Regeneron的pozelimab(C5单抗)联合使用,形成"上游沉默+下游阻断"的双重抑制策略。补体介导疾病市场2025年全球规模约$77亿,预计2034年将增长至$228亿。Alnylam的差异化优势在于:①皮下注射vs.竞品需静脉输注;②siRNA+抗体联合方案可实现更深更持久的补体抑制;③覆盖PNH、重症肌无力、地图状萎缩(GA)三大适应症,市场空间叠加。
✅ Cemdisiran竞争优势
皮下注射(每月一次),vs Soliris/Ultomiris需静脉输注(每2–8周)
siRNA+抗体(pozelimab)联合方案实现"上游沉默+下游阻断"双重抑制
肝源性C5沉默可能比外周C5阻断更完全(覆盖肝脏内C5池)
覆盖PNH/MG/GA三大适应症,特别是GA(眼科)为独有竞争力
与Regeneron合作,pozelimab已获批PNH,联合方案有已验证基础
Phase 2 IgA肾病数据:24小时尿蛋白/肌酐比降低37%,疗效有竞争力⚠️ 主要挑战与竞争
Ultomiris已建立市场主导(每8周输注),医生处方路径依赖
诺华Iptacopan(口服补体B因子抑制剂)的口服便利性是最大差异化威胁
gMG面临C5补体药物的黑框警告,但程度可能比C5单抗轻
Apellis Pegcetacoplan在GA领域已有先发优势
补体疾病市场极度细分——不同疾病中C5抑制效果差异大
联合方案(Cemdisiran+Pozelimab)的定价和医保准入复杂度高
6.4 其他产品竞争格局
7.2025年经营情况与资产负债表分析7.1 2025年收入明细
Alnylam 2025财年实现总收入约$35.3亿(GAAP),同比增长约42%,主要受益于AMVUTTRA在ATTR-CM适应症的持续放量及合作里程碑收入的确认。
收入项目
2025A收入
(百万美元)
同比增速
备注AMVUTTRA(Vutrisiran)
$2,310
+56%
ATTR-CM适应症快速放量,美国市场为主要贡献;2025年6月FDA正式批准ATTR-CM适应症后处方量加速GIVLAARI(Givosiran)
$310
+14%
AHP市场渗透率持续提升,美国以外市场增长稳健OXLUMO(Lumasiran)
$190
+12%
PH1患者诊断率提升,儿童用药占比增加ONPATTRO(Patisiran)
$170
-36%
持续受AMVUTTRA处方转换影响,hATTR-Poly患者向Vutrisiran迁移产品净收入合计
$2,980
+34%
AMVUTTRA驱动增长合作收入
$553
+8%
罗氏Zilebesiran合作里程碑付款、Regeneron/Cemdisiran合作收入等特许权使用费收入
$288
+66%
含Leqvio(诺华)版税$2.28亿、Qfitlia/Fitusiran(赛诺菲)版税$750万、其他版税$0.52亿,合计$2.34亿
总收入
$3,821
+47%
首次全年盈利的里程碑之年
7.2 利润表简要分析
2025年是Alnylam的首个全年GAAP盈利年,标志着公司从Biotech研发型向商业化盈利型企业的关键转折。
利润表项目
2025A
(百万美元)
2024A
(百万美元)
同比变化
总收入
$3,707
$2,607
+42%
减:产品销售成本
($855)
($665)
+29%毛利润
1,464
2,013
3,151
R&D
1,033
1,126
1,320
SG&A
822
976
1,211
其他
$30
($25)
—GAAP税前利润$512($98)扭亏
所得税
($198)
$28
—
GAAP净利润
$314
($70)
7.3 资产负债情况
截至2025年末,Alnylam资产负债表健康,现金储备充裕,为后续管线开发提供了坚实的财务基础。
资产负债表项目
2025年末
(百万美元)
2024年末
(百万美元)现金及有价证券$2,910$2,840
应收账款
$520
$380
存货
$185
$140
固定资产净值
$680
$520总资产$5,680$4,850
短期负债
$620
$580
长期负债(可转债为主)
$1,780
$1,800总负债$2,400$2,380
股东权益
$3,280
$2,470
8.未来5年财务预测(2026E–2030E)
8.1 预测假设
① AMVUTTRA在ATTR-CM患者中持续快速放量,2026E基于公司$44–47亿TTR收入指引建模;
② Nucresiran预计2028年底获批,补充TTR产品矩阵;
③ Zilebesiran预计2028–2029年获批,高血压市场渗透率保守假设;
④ 罕见病产品(Givlaari、Oxlumo)维持稳健个位数至低双位数增长;
⑤ 合作收入假设Leqvio特许权使用费随诺华销售持续提升;
⑥ 毛利率受产品组合和版税结构影响,维持82–84%水平;
⑦ R&D占收入的30%,SG&A占比25%。8.2 产品收入预测详细分析(美国 vs 美国以外)(1) AMVUTTRA(Vutrisiran)收入预测
参数
美国
美国以外
全球合计
数据来源/假设ATTR-CM潜在患者数
~12万确诊(漏诊率~80%)
~25万确诊
~37万确诊
JACC 2024流行病学研究ATTR-PN潜在患者数
~3,000人
~10,000人
~13,000人
遗传性ATTR-PN全球患病率年治疗费用(WAC)
~$50万/年
~$30–40万/年(价格较低)
加权~$45万/年
每季度注射$12.6万×4=$50.3万治疗渗透率(2025E)
~18%
~10%
~14%
参考辉瑞Tafamidis 2019-2024渗透率增长趋势(年均+2-3pp),2025年AMVUTTRA CM获批仅1年治疗渗透率(2028E)
~28%
~18%
~23%
无创诊断推广+医生教育,年增3-4pp(参考历史速率上限)治疗渗透率(2030E)
~35%
~25%
~30%
核素扫描普及+AI辅助诊断,但受限于老年患者诊断意愿和医疗资源AMVUTTRA市占率(在治患者中)
~35%→45%
~25%→40%
加权~38%
Tafamidis先发优势+口服便利,AMVUTTRA需逐步替代;2030E含Acoramidis分流2025A实际收入
~$16.2亿
~$6.9亿$23.1亿
公司财报,US约占70%2026E预测
~$28亿
~$10亿~$38亿
CM渗透率提升+PN维持,增速参考公司指引$44-47亿TTR总收入2027E预测
~$33亿
~$14亿~$47亿
渗透率持续提升,海外加速2028E预测
~$35亿
~$17亿~$52亿
接近峰值,Nucresiran开始分流新患者2029E预测
~$33亿
~$17亿~$50亿
Nucresiran分流+Tafamidis/Acoramidis竞争,存量患者稳定2030E预测
~$31亿
~$17亿~$48亿
Nucresiran持续分流,AMVUTTRA转为"存量维持"模式(2) Zilebesiran收入预测——高血压市场(与罗氏合作)Zilebesiran收入可以参考Inclisiran,二者人群数量相当,但Zilebesiran在难治性高血压的竞争格局预计会比Inclisiran在高血脂上的竞争格局好,后者面临单抗的先发优势。Inclisiran2020年底欧盟获批, 2021 年底美国获批、2023 年中国获批;2021 年销售额 0.12 亿美元,2022 年 1.12 亿美元,2023 年 3.55 亿美元,2024 年 7.54 亿美元,2025 年约 11.98 亿美元,逐年快速增长。市场预测Inclisiran销售峰值30-40亿美元。罗氏预测Zilebesiran销售峰值在30亿瑞士法郎,合33亿美元,个人感觉可能比较保守。如果33亿美元,美国市场占60%(ALNY占50%),美国以外占40%(假设ALNY分15%),对应ALNY收入约12亿美元。(3) Cemdisiran收入预测——补体介导疾病(与Regeneron合作)
参数
PNH
重症肌无力
地图状萎缩
目标患者数(全球)
~6,000人
~60,000人
~150,000人(美国)
年治疗费用
~$20万/年
~$20万/年
~$8万/年
峰值渗透率
~80%
~60%
~60%
Cemdisiran市占率
~20%
~10%
~40%
峰值销售额
~$2亿
~$14亿
~$29亿
ALNY收入
(15%分成)
~$0.3亿
~$2.2亿
~$4.4亿GA适应症,Cemdisiran眼球玻璃体内三个月给药一次,比其他产品2个月或者1个月给药一次具有较大的便利性。三个适应症不确定是否能够实现价格区分。在美国也许能够通过返利实现。gMG适应症,Cemdisiran存在脑膜炎球菌感染潜在风险(所有系统性 C5 抑制剂共性),需提前疫苗接种预防。(4)Inclisiran/Leqvio特许权使用费(诺华商业化)
2025A
2026E
2027E
2028E
2029E
销售额
$12亿
$22亿
$25亿
$28亿
$30亿
ALNY分成
14.5%
15.5%
16.0%
16.5%
17.0%
ALNY收入
$1.74亿
$3.41亿
$4.00亿
$4.62亿
$5.10亿(5)Qfitlia/Fitusiran特许权使用费(赛诺菲商业化)
2026E
2027E
2028E
2029E
销售额
~$3亿
~$6亿
~$8亿
~$10亿
ALNY分成
~18%
~20%
~22%
~24%
ALNY收入~$7,200万~$1.20亿~$1.76亿~$2.40亿8.4 产品收入预测汇总(单位:百万美元)
各产品收入预测(2023A–2030E,单位:百万美元)
收入项目
2025A
2026E
2027E
2028E
2029E
2030EAMVUTTRA
2,314
4,450
4,700
5,200
5,000
4,800
ONPATTRO
173
100
60
30
15
10
GIVLAARI
308
355
400
440
470
490
OXLUMO
191
220
250
275
295
310
现有产品收入
2,9875,1255,4105,9455,7805,610
Nucresiran三代TTR
—
—
—
300
1,400
3,000
Zilebesiran
—
—
—
—
160
500
里程碑收入
553
200
200
200
400
600
特许权收入
174
314
563
870
1138
1400
总收入
3,714
5,639
6,223
7,375
8,878
11,110
8.5 利润率与净利润预测(单位:百万美元)
P&L预测(2023A–2030E,百万美元,GAAP基础)
项目
2025A
2026E
2027E
2028E
2029E
2030E
总收入
3,828
5,639
6,173
7,375
8,878
11,110
成本
677
1,015
1,111
1,317
1,509
1,778毛利润
3,1514,6245,0625,9987,3699,332毛利率
82.3%82.0%82.0%82.0%83.0%84.0%
R&D
1,320
1,692
1,852
2,194
2,663
3,333
R&D占比
34.5%
30.0%
30.0%
30.0%
30.0%
30.0%
SG&A
1,211
1,410
1,543
1,829
2,219
2,778
SG&A占比
31.6%
25.0%
25.0%
25.0%
25.0%
25.0%经营利润
6201,5221,6671,9752,4873,221经营利润率
16.2%27.0%27.0%27.0%28.0%29.0%
其他
100
120
130
140
150
160
所得税
320
246
270
317
396
507
净利润
4001,3961,4271,7982,2412,874
净利润率
10.5%
24.8%
24.7%
24.6%
25.2%
25.9%
注:毛利率分母为产品净收入;整体毛利率使用总收入计算。税率假设2026E–2030E统一按15%测算(基于美国企业税21%及各项税收抵免、爱尔兰知识产权盒等全球税收筹划后有效税率)。SG&A费用率按25%测算,R&D费用率按30%测算,均基于公司持续推进多产品商业化及7个三期临床项目并行推进的战略投入需求。
8.6 关键财务比率汇总
指标
2025A
2026E
2027E
2028E
2029E
2030E
收入增速
65%
47.3%
9.5%
18.5%
21.4%
25.2%
毛利率
82.3%
82.0%
82.0%
82.0%
83.0%
84.0%
R&D费率
34.5%
30.0%
30.0%
30.0%
30.0%
30.0%
SG&A费率
31.6%
25.0%
25.0%
25.0%
25.0%
25.0%
经营利润率
16.2%
27.0%
27.0%
27.0%
28.0%
29.0%
净利率
10.5%
24.8%
24.7%
24.6%
25.2%
25.9%
9.总结与风险分析
9.1短期催化剂AMVUTTRA持续放量
:2026年TTR指引$44–47亿,市场份额快速扩张Qfitlia商业化加速
:赛诺菲推进fitusiran全球上市,特许权使用费提升Cemdisiran三期数据读出
(PNH/MG/GA):2026–2027年Leqvio放量改善
:诺华ORION-4心血管结局数据支撑支付方准入Non-GAAP EPS持续超预期
,强化盈利能力叙事9.2长期价值所在Zilebesiran上市
:若ZENITH三期成功,打开$200亿高血压市场Nucresiran获批
:半年一次注射,强化TTR市场领导地位Mivelsiran CAA/AD数据
:神经科学突破具有重大价值重估潜力MASH/肥胖症读数
:高度竞争赛道,但超大市场机会新一轮授权合作
:AI赋能(Inceptive)加速新靶点发现9.3主要投资风险提示
风险类型
具体风险
概率
影响程度
缓释因素商业风险
AMVUTTRA放量速度低于预期(辉瑞Tafamidis/BridgeBio竞争加剧)
中等
高
HELIOS-B死亡率数据优越,PN+CM双适应症护城河研发风险
Zilebesiran ZENITH三期失败(无MACE获益)
低-中
高(但非核心收入来源)
Phase 2血压降低数据强劲,中间终点已达标监管风险
IRA药价谈判对AMVUTTRA定价施压
中高
中
孤儿药保护,患者数量可能低于谈判门槛技术风险
肝外递送技术突破延迟(神经科学管线风险)
高
中(CNS适应症)
肝靶向业务基础稳固,神经线属于期权价值专利风险
核心专利到期,通用药竞争
低(2030s后)
高(长期)
ESC+化学及序列专利延续保护至2035+
10.4总结
Alnylam(ALNY)是全球 RNAi 小核酸药物绝对龙头,TTR业务持续放量让ALNY有了现金流的托底与公司价值的下限保障。个人直觉判断,仅TTR业务的现金流,就差不多值目前的市值。另一方面,从公司历史股价走势看,近期持续下跌,已经快接近公司历史上最大回撤幅度了,是一个不错的机会。但问题在于公司未来持续的高强度研发投入(公司在业绩交流会上的口径是非GAAP研发费用率达到30%),会较大幅度侵蚀公司账面净利润,让公司的估值持续依赖于未来的研发成功,而不是锚定在市盈率上。
⚠️ 免责声明:本报告仅供研究交流目的,不构成任何形式的投资建议。本报告中的财务预测含有较多不确定性假设,与实际结果可能存在重大差异。投资者应独立进行专业的投资分析并自行承担投资风险。报告编制日期:2026年6月。
临床3期siRNA上市批准突破性疗法临床2期
100 项与 Nucresiran 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 转甲状腺素蛋白介导的淀粉样变性 | 临床3期 | 塞浦路斯 | 2025-12-12 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 美国 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 美国 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 中国 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 日本 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 日本 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 阿根廷 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 阿根廷 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 澳大利亚 | 2025-07-02 | |
| 转甲状腺素蛋白淀粉样变性心肌病 | 临床3期 | 澳大利亚 | 2025-07-02 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
临床1期 | 48 | 鹽製鬱窪夢餘艱夢糧襯(網鏇醖遞鏇糧夢獵衊鏇) = All doses of nucresiran have been well tolerated to date. 淵簾壓艱艱願築艱鹹積 (積蓋蓋襯憲膚窪衊顧積 ) | 积极 | 2024-11-17 | |||

登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

特殊审评
只需点击几下即可了解关键药物信息。
登录
或

芽仔
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用