预约演示
更新于:2026-04-01
Dacinostat
达诺司他
更新于:2026-04-01
概要
基本信息
在研机构- |
权益机构- |
最高研发阶段终止临床1期 |
首次获批日期- |
最高研发阶段(中国)- |
特殊审评- |
登录后查看时间轴
结构/序列
分子式C22H25N3O3 |
InChIKeyBWDQBBCUWLSASG-MDZDMXLPSA-N |
CAS号404951-53-7 |
关联
100 项与 达诺司他 相关的临床结果
登录后查看更多信息
100 项与 达诺司他 相关的转化医学
登录后查看更多信息
100 项与 达诺司他 相关的专利(医药)
登录后查看更多信息
112
项与 达诺司他 相关的文献(医药)2026-02-01·Advanced Science
EIF1AX Nucleolar Condensates Enhance Susceptibilities for the Management of Endometrial Cancer
Article
作者: Chengyu Lv ; Liangzhi Cai ; Qibin Wu ; Shie Wang ; Dabin Liu ; Yuhong Ye ; Pengming Sun ; Jiandong Sun ; Zihang Lin
Abstract:
Endometrial cancer harboring TP53 aberrations presents a significant therapeutic challenge due to the lack of druggable targets. A promising strategy involves inducing senescence in cancer cells followed by targeted elimination using senolytic agents. The preliminary findings indicated that the aberrant subcellular localization of EIF1AX in endometrial cancer is significantly correlated with a poor prognosis. In this study, a compound library is employed to screen for therapeutic agents that induce the nuclear localization of EIF1AX in endometrial cancer cells, followed by a CRISPR library screen to identify senolytic compounds. The results demonstrated that the combination of 2,5‐MeC and dacinostat effectively inhibited tumor growth. Mechanistically, co‐immunoprecipitation mass spectrometry and cleavage under targets and tagmentation sequencing analyses demonstrated that 2,5‐MeC acts as a potent inducer of EIF1AX nucleolar translocation. This translocation promoted senescence by recruiting DDX21 to form nucleolar aggregates, which suppressed rDNA transcription. Additionally, RNA sequencing and antibody array analyses revealed that the synthetic lethality of 2,5‐MeC and dacinostat is mediated through the activation of the JNK/MAPK signaling pathway. Collectively, these findings highlight a novel therapeutic strategy for TP53‐aberrant endometrial cancer.
2025-10-01·Anti-Cancer Agents in Medicinal Chemistry
Redefining Anthraquinone-based Anticancer Drug Design through Subtle Chemical Modifications
Review
作者: De, Utpal Chandra ; Mishra, Bijayashree ; Acharya, Pratap Chandra
Anthraquinones are well known for their wide spectrum of pharmacological properties. Anthraquinone
antibiotics, such as doxorubicin, daunorubicin, epirubicin, and mitoxantrone, have long been used in the clinical
management of various tumors. However, their use is limited due to their toxicity effects, especially cardiomyopathy,
despite their pronounced therapeutic effects. In recent years, medicinal chemists have explored the possibility
of modifying the anthraquinone ring appended with structurally diverse functionality in order to develop
better chemotherapeutic agents with fewer adverse effects. The fused polycyclic structure of anthraquinone offers
rigidity, planarity, and aromaticity, which helps in double helix DNA intercalation, disruption of G4 DNA, and
inhibition of topoisomerase-II enzyme of cancer cells, making them suitable pharmacophore for anticancer drug
discovery. Incorporation of suitable functional groups such as amino, hydroxyl, and their derivatives into anthraquinone
rings can improve their interactions with biological targets involved in cancer progression. These subtle
structural changes produce newer anthraquinone derivatives with improved anticancer properties, increased potency,
selectivity, and reduced toxicity, and can overcome multi-drug resistance. On the other hand, the molecular
hybrids of the anthraquinone derivatives have been reported to act on multiple targets in cancer cells, as seen in
the case of clinical candidates like alectinib, midostaurin, tucatinib, belinostat, and dacinostat. Molecular hybrid
has given a new direction for anticancer drug development, which can produce bifunctional drug candidates with
reduced toxicity. This review summarizes different structural modifications that have been made to the anthraquinone
ring in the last decade with the aim of bringing out potent yet toxicity-free anticancer agents.
2025-09-01·PROTEIN AND PEPTIDE LETTERS
PLEKHG7 Expression: A Biomarker for Prognosis and Targeted Therapy in Diffuse Large B-cell Lymphoma
Article
作者: Lyu, Guizhen ; Li, Dongbing
Introduction::
Pleckstrin homology and RhoGEF domain-containing G7 (PLEKHG7) is
a largely uncharacterized gene whose role in diffuse large B-cell lymphoma (DLBCL) remains unexplored.
Thus, we aimed to profile PLEKHG7 expression, assess its prognostic value, and explore
therapeutic implications.
Methods::
RNA-seq data from TCGA-DLBCL (n=48) and GTEx normal tissues were analyzed via
UCSC XENA. Differential expression was tested using the Wilcoxon rank-sum test and FDR correction.
Prognostic significance was evaluated by Kaplan–Meier and multivariate Cox regression
(nomogram). Gene set enrichment analysis (GSEA) mapped PLEKHG7-associated pathways.
Drug sensitivity correlations were extracted from RNAactDrug. qRT-PCR validated expression in
DLBCL cell lines (OCI-Ly3, SU-DHL-4) versus normal B lymphocytes (GM12878).
Results::
PLEKHG7 was markedly up-regulated in DLBCL tissues (P < 0.001) and cell lines versus
normal controls (AUC = 0.739). High PLEKHG7 expression predicted inferior overall survival
(HR = 8.88; 95% CI: 1.09–72.27; P = 0.041) and remained an independent prognostic factor
(HR = 10.109; P = 0.033). GSEA linked PLEKHG7 to ribosome, oxidative phosphorylation, proteasome,
cytokine-cytokine receptor interaction, spliceosome, and ECM-receptor pathways. Elevated
PLEKHG7 negatively correlated with sensitivity to idelalisib, omipalisib, belinostat,
methotrexate, and dacinostat.
Discussion::
The study's limitations include reliance on bioinformatics data and the lack of functional
validation. Further research is needed to elucidate the molecular mechanisms underlying
PLEKHG7's role in DLBCL and validate its clinical utility.
Conclusion::
PLEKHG7 is significantly overexpressed in DLBCL and independently predicts
poor prognosis. Its association with key oncogenic pathways and drug resistance underscores its
potential as both a prognostic biomarker and a therapeutic target, warranting further functional
validation.
4
项与 达诺司他 相关的新闻(医药)2026-03-30
·今日头条
3月29日,复旦大学附属浦东医院/复旦大学附属肿瘤医院研究团队合作共同在期刊《Advanced Science》上发表了研究论文,题为“DMAP1 Deficiency Suppresses Lung Cancer Progression by Destabilizing Replication Fork and Activating IFN Signaling-Mediated Anti-tumor Immunity”,本研究中,研究人员发现 DMAP1是肺癌进展的关键调节因子。功能研究表明,DMAP1 缺乏通过抑制肿瘤细胞增殖和激活 T 细胞介导的适应性抗肿瘤效应发挥其抗肿瘤作用。临床数据分析显示,DMAP1 高表达与肺癌的“冷”肿瘤微环境和较差的总体生存率相关。
这些发现显著推进了研究人员对 DMAP1 在肺癌发展中的功能的认识,并为设计新的治疗方案提供了科学依据。
肺癌治疗困境与新策略的迫切需求
01
肺癌仍是全球癌症相关死亡的首要原因。在其亚型中,非小细胞肺癌(NSCLC)约占 85%的病例,并在治疗方面取得了显著进展,包括靶向治疗和免疫检查点阻断(ICB)的发展。然而,尽管小分子抑制剂和 PD-1/PD-L1 阻断在某些患者群体中取得了临床成功,但其总体疗效仍有限。许多患者要么最初无反应,要么最终产生耐药性,导致长期预后持续不佳。因此,肺癌患者的五年生存率仍低得令人无法接受,这凸显了寻找新的治疗靶点和策略以提高免疫反应性的迫切需求。
达西诺司他被确认为一种 Dmap1 抑制剂,具有体内免疫介导的抗肿瘤作用
02
鉴于 Dmap1 可能作为抗肿瘤治疗的潜在弱点,研究人员试图通过药物模拟 Dmap1 敲低来重现其抗肿瘤免疫效应。研究人员使用 RNA-seq 数据中上调和下调的前 150 个差异表达基因进行 L1000 预测,并选择了 13 种已进行临床试验或获得 FDA 批准的化合物作为候选药物进行进一步实验验证。
此外,研究人员使用 KP-1 皮下肿瘤模型进行了治疗研究。研究人员将 KP-1 细胞皮下接种到免疫功能正常的 C57BL/6 小鼠体内,当肿瘤体积达到约 100 立方毫米时开始使用达西诺司他进行治疗。治疗组的小鼠每天接受 5 毫克/千克浓度的达西诺司他腹腔注射,而对照组的小鼠则接受溶剂处理。尤其是,达西诺司他治疗显著降低了 KP-1 荷瘤小鼠的肿瘤体积。因此,达西诺司他在体内显著抑制了肿瘤的进展。对免疫微环境的流式细胞术分析表明,达西诺司他促进了 CD4+ 和 CD8+ T 细胞的浸润,这一表型与 Dmap1 敲低时观察到的一致。这些结果表明,达西诺司他作为一种 Dmap1 抑制剂,具有抗肿瘤免疫效应,并凸显了其未来临床应用的潜力。
研究共同表明,非小细胞肺癌(NSCLC)细胞中 DMAP1 缺乏会引发 DNA 复制压力,并通过细胞内在的 I 型干扰素信号激活抗肿瘤 T 细胞免疫,从而揭示了此前未被认知的表观遗传 - 免疫轴,并凸显 DMAP1 为潜在的治疗弱点。
Dmap1 缺失阻碍肺癌进展的工作模型
结论
03
总之,本研究揭示了 DMAP1 在肺癌中出人意料的致癌作用,确立了其此前未被认识到的功能,并阐明了抗肿瘤免疫之间的机制联系。此外,本研究工作凸显了靶向依赖 DMAP1 的复制和免疫通路的具有治疗可行性的策略。
参考资料:
https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202517634
【关于投稿】
转化医学网(360zhyx.com)是转化医学核心门户,旨在推动基础研究、临床诊疗和产业的发展,核心内容涵盖组学、检验、免疫、肿瘤、心血管、糖尿病等。如您有最新的研究内容发表,欢迎联系我们进行免费报道(公众号菜单栏-在线客服联系),我们的理念:内容创造价值,转化铸就未来!
转化医学网(360zhyx.com)发布的文章旨在介绍前沿医学研究进展,不能作为治疗方案使用;如需获得健康指导,请至正规医院就诊。
责任声明:本稿件如有错误之处,敬请联系转化医学网客服进行修改事宜!
微信号:zhuanhuayixue
2024-10-02
摘要:抗体-药物偶联物(ADCs)是一种快速发展的靶向生物治疗药物类别,目前结合了单克隆抗体的选择性和由细胞毒素组成的有效载荷的效力。多年来,微管靶向和DNA嵌入剂一直是ADC开发的核心。最近,两种基于拓扑异构酶1抑制剂的ADC——曲妥珠单抗德鲁替康(Enhertu®)和萨奎图珠单抗戈维替康(Trodelvy®)的批准和临床成功,展示了将非传统有效载荷与差异化作用机制结合的潜力。在未来ADC领域的发展中,有效载荷多样化预计将发挥关键作用,这一点从越来越多的临床前和临床阶段非传统有效载荷偶联ADC的数量中可以看出。本综述提供了一个全面的概述,涵盖了不同作用机制的经过验证的、被遗忘的和新开发的载荷。
1.引言
使用单克隆抗体作为治疗载体是Paul Ehrlich在“神奇子弹”概念中提出的假设。生物学和化学方面的显著进步导致了抗体-药物偶联物(ADCs)的开发,这是新一代的生物治疗药物,结合了抗体的高度特异性和小分子细胞毒素的效力,目的是在目标细胞内传递高度有效的有效载荷。这些武装化的抗体在某些情况下可以显著提高细胞毒素分子的治疗指数,并减少它们的非靶向毒性,这是传统细胞毒素化疗的主要问题。
抗癌ADCs由三部分组成:一种特异性识别目标细胞上抗原的单克隆抗体(mAb)、一种在释放时触发细胞死亡的有效小分子,以及连接这两个元素的链接器。
这些药物的复杂性解释了它们艰难的开发过程,最好的例子是gemtuzumab ozogamicin的混乱历史,它最初在2000年获得批准,在2010年从大多数市场撤出,然后在2017年被FDA重新批准。在mAb设计、目标选择、偶联技术、有效载荷选择和质量控制标准化方面的主要进步,使这个家族演变成了抗癌药物的成熟组成部分,目前有13种药物获得批准用于治疗癌症,其中7种是在过去三年中获得批准的。
鉴于有效载荷的效力,抗体必须对其目标高度选择性,并在偶联后保持其半衰期和生物学特性。抗体工程,如Fc(可结晶片段)沉默或Fc容量增强,是一种强大的工具,分别用于平衡由T细胞靶向引起的抗体非靶向毒性或增加抗体功能,如ADCC和ADCP,两种方法都已在治疗性mAbs的背景下证明其临床益处。在ADCs的背景下,假设Fc沉默将显著减少非靶向毒性。血小板减少症和中性粒细胞减少症是接受ADCs治疗的患者常见的不良反应,可能与血小板表面FcγRIIa受体的表达有关。MEDI4276,一种减少了FcγR结合的曲妥珠单抗艾美坦辛(T-DM1,Kadcyla®)的类似物,已在临床试验中进行了研究,旨在减少与T-DM1观察到的血小板减少症(NCT02576548)。令人惊讶的是,这种ADC在首次人体试验中显示出显著的毒性。相比之下,Fc容量增强已通过brentuximab vedotin(Blenrep®)的批准证明了其益处,其mAb是岩藻糖基化的。最近mAb衍生物的开发扩大了载体的可能性,同时旨在改善ADC载体的基本特性。较小的格式,如mAb片段(scFv、单链可变片段和Fabs、抗原结合片段),在这种情况下进行了探索(小格式药物偶联物),目的是增强实体肿瘤的穿透和细胞内化。然而,到目前为止,这些候选药物没有进入临床试验,因为它们被发现面临快速消除,可能因此无法提供比传统mAb格式的好处。相比之下,另一类基于自行车肽的小格式药物偶联物似乎满足了与mAb格式偶联物竞争的挑战,具有竞争性的摄取效率,正如目前正在进行的三个自行车肽偶联物的临床试验所说明的(NCT04561362、NCT04180371和NCT03486730)。
除了大小之外,其他重要参数影响这些大分子的药代动力学(PK)和循环半衰期,包括化学修饰,影响它们通过肾小管上皮细胞的保留,以及它们通过新生儿Fc受体(FcRn)的回收率。小格式偶联物可能受益于这些参数的调节。多价结合实体,如二价抗体或双特异性抗体,正在开发中,以提高抗原亲和力、选择性或内化,并可能构成有前途的载体。
最广泛应用的生物偶联方法使用赖氨酸侧链胺和半胱氨酸链间硫醇。然而,所得混合物的异质性一直是ADC失败的主要问题。其他偶联策略已经开发出来,包括通过特定或工程化氨基酸的位点特异性偶联,但迄今为止未能在临床试验中证明改善结果。Iladatuzumab vedotin(DCDS0780A),一种FDA批准的polatuzumab vedotin(Polivy®)的THIOMAB™版本,在临床试验中进行了探索,但由于在测试剂量下的过度眼部毒性而未能进入II期。这种THIOMAB™药物偶联技术展示了巨大的潜力,但两个mAbs之间的区别和/或在临床设计上,包括选定的剂量、适应症和患者群体,可能导致一个获得批准而另一个没有。在这种情况下,重要的是要强调不仅技术很重要。正在探索新的偶联策略,如糖链和短肽标签(酶辅助连接)或最近通过ADP-核糖环化酶,目的是生成均匀且物理稳定的ADCs。
连接子在抗体药物偶联物(ADC)设计中扮演着重要角色,因为它强烈影响着ADC的安全性、效力和活性。最重要的是,连接子需要在循环中保持稳定,以避免药物过早脱落,同时允许其在目标细胞内释放。根据其可切割性,已经开发并区分出两类连接子。可切割连接子要么对pH敏感,如腙连接子,要么对谷胱甘肽或二硫化物异构酶敏感,如二硫键连接子,要么对蛋白酶敏感,如二肽键对卡他普辛B。不可切割连接子依赖于抗体部分的溶酶体降解,从而至少保留一个氨基酸,通常是赖氨酸或半胱氨酸,连接到载荷-连接子复合物。这种方法提高了连接的稳定性,因为需要抗体消化才能释放载荷。虽然强调连接子越稳定,引起的非靶向毒性就越少,但这些技术通常发现过于严格,无法支持抗肿瘤活性。ADC的安全性仍然是其设计中的主要挑战,靶向和非靶向毒性不仅由连接子-载荷的不稳定性驱动。实际上,靶向毒性总是由单抗及其对目标的亲和力/亲和力驱动,以及载荷的作用机制,这再次说明了肿瘤类型、目标抗原和ADC结构匹配的重要性。最近对连接子设计改进的兴趣导致了亲水性连接子的开发,以平衡载荷的疏水性。硫酸盐、聚乙二醇(PEG)、多唾液酸(PSAR)或最近基于DNA的连接子显著提高了ADC的稳定性和药代动力学,导致毒性更低、活性更高的ADC。
药物-抗体比率(DAR)直到现在一直保持在四个以下,以避免单抗聚集并限制ADC的整体疏水性,这与毒性、缩短的半衰期和狭窄的治疗指数相关。因此,增加DAR更适合于疏水性较低或得到良好补偿的疏水载荷,如新型连接子技术所示,包括掩蔽实体或连接子亲水插入物,其效率直接决定了DAR增加的能力。恢复亲水性和裸mAb药代动力学特性以及高DAR显著增加了载荷对肿瘤的暴露。这些新的连接子技术使得开发比DNA插入或微管干扰剂更少效力的载荷成为可能,如两个基于拓扑异构酶1的ADC在DAR8处的结合所示,曲妥珠单抗deruxtecan(Enhertu®)和sacituzumab govitecan(Trodelvy®)。这些高度装载的ADC与非传统载荷也可能潜在地扩大ADC的治疗适应症,通过解决高或低目标表达水平的肿瘤,如基于DESTINY-Breast04结果最近批准的Enhertu®在HER2低表达肿瘤中所示。相反,在高效力载荷的背景下,到目前为止,较低的DAR更受欢迎,如最近批准的loncastuximab tesirine(Zynlonta®),一个DAR2的极强吡咯苯并二氮杂卓(PBDs)载荷。值得注意的是,与在临床试验中评估的曲妥珠单抗deruxtecan和类似物在DAR8处结合不同,datopotamab deruxtecan的DAR降低到4,以减少其目标驱动的毒性,这表明了ADC设计的多维性。
载荷(也称为弹头)发挥了ADC的细胞内细胞毒性活性。通过连接子部分共价结合到抗体的细胞毒素的性质非常重要,因为它的作用机制将决定ADC作为抗癌化合物的效力及其可能的适应症。第一代ADC结合了传统化疗药物(紫杉醇、蒽环类),因为载荷不够有效,只有一小部分总偶联物成功地在目标细胞内传递了它们的载荷。肿瘤穿透、细胞表面的靶标拷贝数以及ADC的内化和降解强烈影响细胞内游离载荷的浓度。因此,载荷必须在低浓度下具有高度效力,其50%抑制浓度(IC50s)在低至亚纳摩尔范围内。其他因素,如分子在血浆中的稳定性和在酸性条件下的稳定性、结合位点的可及性或溶解度至关重要。多年来,载荷基本上由两类组成:微管抑制剂,包括美登素和auristatins,以及DNA烷基化剂,如calicheamicins。这些载荷导致了八个ADC的批准(图1)。
图1 FDA对抗癌ADCs的批准。根据其有效载荷的性质对ADCs进行识别:微管破坏剂;DNA靶向剂:卡利奇霉素,吡咯并苯并二氮杂环(PBD),拓扑异构酶1(TOPO 1)抑制剂。
新型且更有效的DNA烷基化剂,如PBD单体和二聚体、indolino-benzodiazepines(IGNs)或cyclopropabenzindolone(CBI)单体和二聚体,IC50值在皮摩尔范围内,一直处于ADC设计的前沿。这些分子是有史以来合成的最强大的抗肿瘤化学物质之一,它们通过ADC结构特异性靶向肿瘤细胞以产生高度有效的“魔法子弹”进行了研究。然而,强烈的剂量限制毒性限制了它们的临床发展,目前只有loncastuximab tesirine在2021年获得了FDA批准(图1)。已经做出了努力,使载荷家族多样化,以获得具有原始作用机制的分子,包括几个不直接针对DNA或微管的分子。效力较低的分子从ADC结构的重大突破中受益,通过改进的连接子设计允许更高的DAR值、更稳定的载荷附着或增强的旁观者杀伤活性。最近和引人注目的成功是拓扑异构酶1(topo-1)抑制剂的开发,这代表了载荷选择的转折点,自2019年以来批准了两个基于topo-1抑制剂的ADC(图1),曲妥珠单抗deruxtecan(Enhertu®,DS-8201a)和sacituzumab govitecan(Trodelvy®)。最近的综述已经描述了ADC的验证和探索性治疗靶标的格局。这篇综述旨在描述ADC背景下验证的、被遗忘的和新开发的载荷的格局,具有多种作用机制,不包括微管抑制剂和DNA烷基化剂。
2.成功的载荷家族:拓扑异构酶1抑制剂
拓扑异构酶1抑制剂是FDA最近批准的抗体-药物偶联物载荷家族,最初由曲妥珠单抗deruxtecan推动,随后是sacituzumab govitecan(表1,图2A)。这些基于中等效力载荷的偶联物的最近发展得益于生产了DAR值为8的高负载ADC。
图2 拓扑异构酶I抑制剂基础的ADCs结构图。A FDA批准的ADCs和有效载荷(紫色)以及正在临床评估中的ADCs和有效载荷(蓝色)。B 用于临床前开发的拓扑异构酶1抑制剂(绿色)。C 作为ADCs潜在有效载荷的下一代拓扑异构酶I抑制剂。图中注释:[ADC名称],抗体,有效载荷。
拓扑异构酶酶位于细胞核内。它们的作用是控制和修复在DNA开放、上游转录和复制过程中发生的DNA超螺旋和纠缠。这些催化酶切割、修复超螺旋并重新连接DNA链。拓扑异构酶根据它们的切割活性分为两个家族:拓扑异构酶I切割单链DNA,而拓扑异构酶II切割双链DNA。拓扑异构酶抑制剂特异性结合到DNA-拓扑异构酶复合物的界面,从而抑制拓扑异构酶修复机制,导致DNA损伤和随后的细胞凋亡。然而,最有效的拓扑异构酶抑制剂的效力比美登素或calicheamicin低100到1000倍,这解释了在最初的ADC设计中对这种载荷类别最初缺乏兴趣。
这种载荷类别包括基于喜树碱和非喜树碱的化合物。喜树碱(CPT)是一种由五个化学环组成的天然植物生物碱,其水溶性差(图2B)。几种衍生物已经获得监管机构的批准,具有改善的生物利用度,即拓扑替康、伊立替康和贝洛替康。这些药物已获得批准用于多种适应症,包括卵巢癌、肺癌、宫颈癌和结直肠癌。伊立替康的脂质体制剂也已获得批准用于治疗晚期胰腺癌。还合成了几种其他CPT衍生物,如gimatecan,目前正在II期评估中,用于治疗卵巢癌、输卵管癌或腹膜癌(NCT04846842)。基于CPT的分子最严重的不良事件(SAEs)包括严重水样腹泻、中性粒细胞减少和血小板减少。
由于其中等细胞毒性效力,CPT衍生物最近被用作ADC载荷,其IC50值在低纳摩尔范围内。它们的效力介于非常有效的(皮摩尔IC50s)抗微管/DNA靶向剂和最初用于最初的ADC项目的传统(微摩尔IC50s)化疗药物之间,后者因缺乏效力而失败(甲氨蝶呤和多柔比星)。到目前为止,有两种CPT衍生物已成功结合到抗体上并获得批准:DXd和伊立替康的活性代谢物SN-38(表1)。
2.1.Exatecan及其衍生物
DXd是exatecan(也称为DX8951f)的衍生物,与CPT相比,该化合物具有增强的活性和改善的溶解性,并且被描述为不是ABCC2或ABCG1的底物。未偶联的exatecan在几项临床试验中进行了评估,但由于其狭窄的治疗窗口,剂量限制性中性粒细胞减少和血小板减少以及强烈的胃肠道毒性,没有提高生存率。最初尝试将exatecan生物偶联到抗体上取得了部分成功,但偶联物出现显著聚集。这个问题通过第一三共制药科学家使用稍微修改的exatecan衍生物,名为DXd,得到了解决。发现这种新化合物保留了exatecan的效力,同时能够成功生物偶联多达8个DXd分子每个抗体,而没有显著的聚集。这种deruxtecan药物连接子在几个专有ADC项目中使用,例如DS-8201a(Enhertu®)、U3-1402和DS-6157a,以DAR8偶联,DS-1062a和DS-7300a以较低的DAR(4)偶联以限制它们的毒性[39],它们要么已经获得FDA批准(DS-8201a),要么目前正在临床评估中(表1,图2A)。尽管DXd载荷展示了比exatecan甲磺酸盐更低的被动膜渗透性,但发现它较少骨髓毒性,因此也被选择用于其改善的安全概况。
曲妥珠单抗deruxtecan(DS-8201a或Enhertu®)由已获批准的靶向HER2的抗体曲妥珠单抗组成,通过一个基于马来酰亚胺的mc-GGFG-am蛋白酶可切割连接子连接到8个DXd载荷(图2A)。这种创新的DAR8 ADC在优化的连接子和载荷方面,与第一代ADC相比,在临床前展现了改善的治疗窗口。在两项大型3期研究(DESTINY-Breast03, NCT03529110, DESTINY-Gastric01, NCT03329690)之后,曲妥珠单抗deruxtecan已于2019年获得FDA批准用于治疗不可切除或转移性HER2+乳腺癌(BC),并于2021年获得批准用于治疗晚期或转移性HER2+胃癌或胃食管癌,2022年稍后用于治疗不可切除或转移性HER2+非小细胞肺癌(DESTINY-Lung02)。值得注意的是,曲妥珠单抗deruxtecan在曲妥珠单抗emtansine治疗后复发的乳腺癌患者中展现了强大的抗肿瘤活性,并在胃癌患者中显示出比伊立替康更强的活性,在非小细胞肺癌(NSCLC)和结直肠癌中显示出持久的抗癌活性。另一个ADC批准的突破是由其在DESTINY-Breast04试验中的临床评估所展示,该试验导致其在2022年获得批准用于治疗不可切除或转移性HER2低表达乳腺癌。目前还有几项其他临床试验正在进行中,包括DESTINY-breast05和DESTINY-breast09,分别评估Enhertu在新辅助疗法后残留疾病HER2+BC患者中的使用或与当前一线标准护理方案在HER2+BC中的对比,再次展示了其成功。其他四个含有这种有前景的连接子-载荷的ADC目前正在进行临床评估,用于治疗实体瘤,分别靶向NSCLC中的HER3、转移性结直肠癌和乳腺癌、TROP2在NSCLC和三阴性乳腺癌(TNBC)、B7-H3在晚期实体瘤或GPR20在胃肠道间质瘤(GIST)中(表1)。
最近,由于开发了能够绕过化合物的疏水性和促聚集特性的亲水性可切割连接子结构,exatecan(图2B)已经在临床前作为潜在的ADC载荷进行了探索。这允许在不干扰ADC药代动力学特性的情况下以高DAR值偶联exatecan(表1)。这些ADC在肿瘤异种移植物中展现了强大的抗肿瘤活性,并由于与DXd相比exatecan改善的被动细胞渗透性,与基于deruxtecan的ADC相比显示出更强的旁观者杀伤效应。目前正在开发两种使用这种药物连接子策略的ADC:PRO1184和PRO1160,分别含有亲水性exatecan基连接子,以DAR8偶联到抗FRa和抗CD70抗体,并预计将于2023年进入临床试验。最近的体内研究还表明,exatecan不需要呋喃环功能就能发挥其抗肿瘤活性,从而扩大了该分子的功能化可能性,以生成可连接的衍生物(表1)。使用这种策略开发的最有前途的ADC(mAbE-21a,衍生物11,DAR7.5)在EGFR+模型中以0.25 mg/kg剂量展现了显著的抗肿瘤活性,完全缓解。
今年披露了一种新型专有exatecan衍生物AZ’0132,正在作为ADC AZD8205的载荷进行研究,靶向B7-H4(表1,图2A)。AZD8205目前正在进行I/II期研究,用于治疗乳腺癌、卵巢癌和子宫内膜癌以及胆管癌(NCT05123482)。
2.2.伊立替康
伊立替康已获得FDA批准用于治疗各种实体瘤,如胃肠道恶性肿瘤、胶质母细胞瘤和宫颈癌,是拓扑异构酶1抑制剂SN-38的前药。SN-38水溶性差,会引起严重的毒性,包括强烈的骨髓抑制和高级别腹泻。因此,开发了伊立替康以改善生物利用度并获得可接受的治疗指数。IMMU-132(Trodelvy®)是一种抗TROP2抗体,与基于SN-38的药物连接子偶联(表1,图2A)。这种ADC已于2020年获得FDA批准用于治疗三阴性转移性乳腺癌和转移性尿路上皮癌,目前正在进行临床试验,用于治疗HR+/HER2-、前列腺和子宫内膜癌(NCT03725761和NCT04251416)。其他ADC也已开发出这种基于SN-38的连接子,包括IMMU-130(labetuzumab govitecan)[84–86]和IMMU-140,分别靶向CEACAM5和HLA-DR(表1)。Labetuzumab govitecan在I期(NCT01270698)中展现了可接受的毒性和活性,然而,II期评估在2020年因未公开原因终止(NCT01915472)。IMMU-140针对HLA-DR,在血液病和黑色素瘤中展现了有希望的临床前活性。SN-38载荷也在临床前评估中针对各种液体肿瘤(表1)。据我们所知,尽管临床前结果有希望,但这些ADC中没有一个进入临床试验,最近的相关出版物超过七年前。最近,为了治疗自身免疫疾病,绕过激素抵抗,开发了一种A7R-SN-38 ADC(表1)。
2.3.Belotecan衍生物
另一种拓扑异构酶1抑制剂KL610023,是FDA批准的分子belotecan的衍生物,正在作为ADC载荷进行研究。这种拓扑异构酶I抑制剂被开发用于生成抗TROP2 ADC(SKB-264),目前正在进行I/II期临床试验(NCT04152499)治疗各种实体瘤的患者(表1,图2A)。
2.4.其他拓扑异构酶1抑制剂
作为ADC载荷的喜树碱衍生物的一个限制是分子内缺乏可连接的化学胺基团。为了在不改变其抗肿瘤性质的情况下在载荷内插入可连接的功能,已经合成了其他CPT衍生物(表1)。在这些衍生物中,cAC10的临床前研究,一种抗CD30抗体偶联到8个AMDCPT分子,显示出非常有希望的结果(图2B)。最近开发了几种非喜树碱衍生物,包括吲哚异喹啉、二苯萘吡啶酮和氟吲哚异喹啉(图2C)。这些分子显示出与CPT衍生物相比具有几个优点,包括更高的细胞毒性、改善的稳定性或延长的活性,并且目前正在作为小分子进行早期临床试验。LMP-517,与氟吲哚异喹啉偶联,正在被研究;然而,据我们所知,还没有数据被披露。
3.已经进入临床试验的载荷:承诺和失败
虽然拓扑异构酶1抑制剂深刻地改变了ADC载荷格局,但其他几个药物已经在临床试验中进行了评估。表2总结了在患者中评估的原始载荷。主要类别包括拓扑异构酶2抑制剂、RNA聚合酶抑制剂、Bcl-xL抑制剂和免疫刺激剂。此外,糖皮质激素现在作为超出肿瘤学适应症的ADC载荷出现了。
3.1.拓扑异构酶2抑制剂
拓扑异构酶2抑制剂在血液病恶性肿瘤和实体瘤的抗癌治疗中广泛使用。它们的机制复杂,可能不仅涉及直接抑制拓扑异构酶2活性,还包括DNA插入、ROS诱导和线粒体破坏。它们的毒性谱包括骨髓抑制、胃肠道毒性,在某些情况下还有高级别心脏毒性。多柔比星作为治疗乳腺癌、膀胱癌和甲状腺癌以及淋巴瘤和多发性骨髓瘤的一线治疗已经使用了几十年。多柔比星也是ADC开发中使用的第一批载荷之一,当时常规化疗小分子首次被偶联。第一个含有拓扑异构酶2抑制剂的ADC(SGN-15, BMS-182248)由多柔比星偶联到靶向Le-Y抗原的小鼠BR-96抗体(表2,图3)。SGN-15是在ADC发现的最初阶段与KS1/4-甲氨蝶呤(表2)一起在20世纪80年代开发的,用于治疗前列腺癌、乳腺癌和NSCLC。它的I期临床试验显示了可接受的耐受性,然而II期由于连接子不稳定和Le-Y靶标在正常组织中的表达导致了非靶向毒性。因此,ADC在可接受的剂量下缺乏效力。已经批准的化疗药物偶联观察到的令人失望的结果形成了共识,即ADC载荷应该比常规化疗药物更强效。这导致了第二代ADC的开发,它们被偶联到更强效的载荷上,如微管抑制剂和DNA损伤剂。
图3 展示了已经进入临床试验的抗体-药物偶联物的结构以及它们有效载荷(蓝色)的分类,这些有效载荷根据它们的作用机制进行了分类。图中注释:[ADC名称],抗体,有效载荷。
尽管早期开发不尽人意,多柔比星后来被偶联到靶向CD74的抗体milatuzumab(IMMU-110)用于治疗多发性骨髓瘤(表2,图3)。这种ADC被带到临床试验中,但显示出令人失望的效力,其开发在2013年被终止(NCT01101594)。此外,多柔比星还被用作ADC载荷在临床前连接子概念验证研究中(表3),它被偶联到一个新的可切割连接子(NEBI)或一个不可切割连接子(SMAC)。SMAC研究的结果表明,不可切割连接子对于基于多柔比星的ADC开发可能过于严格,因为没有观察到细胞毒性。
鉴于多柔比星作为ADC载荷的效力不足,另一种蒽环类药物PNU-159682,其细胞毒性是多柔比星的100倍,后来被探索。除了比其他拓扑异构酶2抑制剂更强效外,PNU-159682不是efux泵的底物。被发现是efux泵底物的载荷是ADC开发中的限制因素。2020年,一种基于PNU-159682的新型ADC,NBE-002(表2,图3),针对ROR1,进入I/II期临床试验(NCT04441099)。有趣的是,NBE-002诱导了长期免疫保护,这表明它可以成功地与免疫检查点抑制剂(ICIs)结合。SOT102(前称SO-N102)是另一种有前景的基于PNU-159682的ADC,靶向CLDN18.2(表2,图3)。SOT102在低表达肿瘤中展示了大的治疗窗口,并已于2022年4月进入I期临床试验(EudraCT编号2021-005,873-25)。PNU-159682的许多临床前用途也被报道,结果证明了它能够绕过常用载荷如MMAE或DM1的抗性机制(表3)[110, 111, 114–118]。PNU-159682载荷还与MMAE一起偶联形成双药ADC(表3)。然而,尽管体外同时观察到了两种作用机制,但没有观察到协同作用。
在20世纪90年代的临床前阶段也开发了柔红霉素和伊达比星的偶联物(表3,图4),但显示出降低的效力。一种抗HER2 afbody-idarubicin偶联物最近在体外进行了评估,具有特异性针对HER2阳性头颈鳞状细胞癌(HNSCC)细胞而不是HER2阳性BC细胞(表3)。
图4 非传统ADC有效载荷的化学结构图,这些有效载荷处于临床前阶段。
3.2.转录抑制剂
转录在细胞发展、活动和增殖中具有基本作用,因此可以构成ADC载荷的一个创新和原始靶标。转录由RNA聚合酶II(RNApolII)调节,它直接与DNA结合,并涉及转录因子,这些因子与RNApolII形成复合体以启动转录(如TFIIH)和共同调节因子(如组蛋白去乙酰化酶,HDAC),这些因子介导染色质结构和可及性。尽管一些HDAC抑制剂已经获得批准,但由于耐受性差,目前还没有批准的RNApolII抑制剂。
毒蝇伞素是从毒蝇伞蘑菇中提取的天然且高效的RNApolII抑制剂。α-毒蝇伞肽和β-毒蝇伞肽连同其他七种大环衍生物构成了毒蝇伞素家族。尽管它们作为实验室试剂广泛用于探索转录机制,但α-毒蝇伞肽被证明毒性太大,特别是对肝脏,以至于不能作为抗癌药物进一步开发。然而,这种分子作为潜在ADC载荷具有许多优点,包括其原始的细胞内靶标、其有利的物理化学性质(包括亲水性)、其对efux泵的不敏感性,以及其在静止癌细胞中产生细胞毒性的能力。相比之下,毒蝇伞肽的亲水性预计将阻止通过旁观者效应杀死邻近细胞,这可能限制其在均匀分布的靶标中的使用。即使在这些情况下,完全缺乏旁观者效应可能导致由于患者之间的靶标分布不同而导致效力不足。
毒蝇伞肽衍生物β-毒蝇伞肽最早在1973年与白蛋白偶联,这种ADC前体证明了选择性杀死巨噬细胞(表3)。后来,这个衍生物被偶联到抗MUC1和抗PSMA抗体上,在临床前模型中展示了强大的选择性细胞毒性(表3)。它的类似物α-毒蝇伞肽及其衍生物偶氮-毒蝇伞肽也很早就被用作ADC载荷(表3)。偶氮-毒蝇伞肽-ADC比未偶联的分子显示出大约500倍的细胞毒性。这可以通过分子的亲水性来解释,这种亲水性降低了细胞膜的渗透性,同时作为ADC结构被有效内化。截至2021年5月,第一个毒蝇伞肽-抗体偶联物(ATAC®)候选药物HDP-101已经进入早期临床试验(表2,图3)。HDP-101是一个针对BCMA的ADC,目前正在评估用于治疗多发性骨髓瘤和浆细胞疾病的患者(NCT04879043)。ATAC最近被描述为免疫激活药物。它们被发现可以诱导免疫原性细胞死亡(ICD),并与ICI表现出协同作用,这为临床环境中的组合可能性开辟了新的视野。值得注意的是,针对其他靶标的一些α-毒蝇伞肽ADC(EpCam、HER2、PSMA、CD19)在体外和体内都显示出强大的抗肿瘤活性(表3)。α-毒蝇伞肽还作为双弹头与MMAE偶联(表3)。这种DAR 1+1 ADC靶向FGFR1,在体外显示出强大的细胞毒性。其他高效的RNApolII抑制剂在20世纪90年代被偶联,如鬼笔环肽,以及霉菌毒素三烯醇、疣状菌素A和roridin A(表3,图4)。考虑到自90年代以来ADC设计的进步,以及这些化合物在各种细胞系中的纳摩尔细胞毒性,这些分子可能在未来几年成为进一步探索的对象。
另一种阻止DNA转录的策略是抑制转录因子(TFs)。TFs对RNApolII附着在DNA上的启动步骤至关重要。TF抑制剂(TFi)已经证明它们在临床试验中具有抗肿瘤活性,其中水溶性前药minnelide目前正在II期评估中(NCT04896073)。雷公藤内酯,一种来自被称为“雷公藤”的中草药的天然化合物,具有高度的细胞毒性,但也具有疏水性,具有较差的生物利用度和高毒性(图4)。因此,正在努力开发具有更好药物化学特性的类似物。另一种策略是将这种分子偶联到靶向实体上,从而绕过这些问题。雷公藤内酯最近首次被偶联到抗CD26抗体上,以靶向间皮瘤和淋巴瘤(表3)。这种不可切割的ADC在目标细胞中有效地停止了mRNA合成,并在体外和体内展示了有希望的抗肿瘤活性。还开发了一种针对EGFR阳性肺癌的cetuximab-triptolide ADC(表3)。这种ADC对EGFR过表达模型具有选择性,并比未偶联的雷公藤内酯具有更低的毒性。Cetuximab-triptolide有效地诱导了转录抑制,在体外和体内都具有强大的抗肿瘤活性。还评估了针对HER2的雷公藤内酯ADC,并取得了类似的结果。然而,对于每种基于雷公藤内酯的ADC,都需要高剂量才能在异种移植模型中观察到抗肿瘤活性,这些论文中没有报告最大耐受剂量,从而质疑了治疗指数的宽度。
组蛋白去乙酰化酶(HDACs)影响转录因子,因此参与包括转录在内的各种细胞过程。它们被发现在癌细胞中过度表达或过度激活,并被认为参与增加增殖、迁移和侵袭。Vorinostat和dacinostat是FDA批准的两个HDAC抑制剂(HDACi)的例子。然而,这些分子呈现出强烈的系统性副作用风险,如血小板减少和胃肠道毒性,以及较差的药代动力学(PK)特性。自2018年以来,它们已经在ADC设计中进行了研究:ST74612AA1是第一个生物偶联的HDAC抑制剂(表3,图4)。这种相对无毒的分子是第二代泛HDACi。这种分子被偶联到cetuximab和trastuzumab上,两种ADC都比未偶联的HDACi具有更安全的特性,同时在细胞系衍生的异种移植(CDX)和患者衍生的异种移植(PDX)模型中都具有活性。然而,正如观察到的基于TFi的ADC一样,异种移植模型接受了30 mg/kg的高剂量治疗。2020年,vorinostat和dacinostat也被偶联到cetuximab和trastuzumab上,在体外具有有趣的抗增殖活性(表3,图4)。
3.3.Bcl-xL抑制剂
Bcl-2家族成员可能是促凋亡蛋白(Bad、Bim、PUMA、Bik、Bak等)或抗凋亡蛋白(Bcl-2、Bcl-xL、Bcl-w、Mcl-1等)。在癌细胞中,这些蛋白之间的平衡通常倾向于生存,使得抗凋亡蛋白成为创新ADC载荷的有趣和原始靶标。
根据它们的化学功能骨架,Bcl-xL和Bcl-2抑制剂被分类为4大家族:N-酰磺胺类(navitoclax、venetoclax)、吲哚类(obatoclax)、棉酚乙酸(AT-101、sabutoclax)和苯并噻唑腙类(如WEHI-539)。Bcl-xL的抑制与严重的血小板减少有关,这证明了寻找高度特异性Bcl-2抑制剂的合理性,如venetoclax。目前venetoclax已获批准用于慢性淋巴细胞白血病的一个亚组患者和急性髓系白血病。
ABBV-155(mirzotamab clezutoclax)是一种抗B7-H3抗体,与Bcl-xL抑制剂clezutoclax偶联(表2,图3)。这种创新ADC于2018年进入正在进行的I/II期临床试验,作为单一药物用于晚期实体瘤的治疗,以及与紫杉醇联合用于晚期非小细胞肺癌和乳腺癌患者(NCT03595059)。在单一药物I期队列中包含的前31名患者中没有报告剂量限制毒性,SAEs包括贫血、淋巴细胞计数下降、疲劳和腹泻。在紫杉醇组合臂中观察到21%的患者部分缓解。
3.4.酪氨酸激酶抑制剂
人类激酶组包含超过500个激酶,其中150多个与各种疾病相关,包括癌症。蛋白激酶是催化磷酸化的酶,分为3类:丝氨酸、苏氨酸或酪氨酸激酶。目前正在临床试验中研究的小分子中超过四分之一是蛋白激酶抑制剂,超过30个FDA批准的癌症治疗分子是激酶抑制剂。在癌症中,各种激酶家族参与细胞周期进展、细胞增殖、运动和血管生成。自2001年首个激酶抑制剂伊马替尼获批以来,激酶抑制剂被分为5类:I型和II型是ATP竞争性,分别针对激酶的活性或非活性形式;III型结合ATP的变构口袋;IV型结合激酶的变构口袋,V型结合多种结合模式。
虽然在癌症治疗中被广泛探索,但蛋白激酶抑制剂作为ADC载荷并未被广泛探索,可能是因为它们的效力较低。抗CD19抗体B43已与染料木素(一种含有大豆异黄酮的植物雌激素)偶联,发现它通过抑制表皮生长因子受体(EGFR),一种酪氨酸激酶受体,诱导凋亡和细胞增殖抑制(表2,图3)。体外和体内的临床前研究(小鼠、大鼠、非人灵长类:NHP)表明在100 mg/kg的累积剂量下没有毒性,并且在小鼠模型中比标准化疗具有更强的抗肿瘤效果。这些有希望的结果导致了它在1999年首次用于治疗ALL和NHL的人体研究。除了在人类中呈现有利的药代动力学特性外,没有报告毒性和有希望的抗肿瘤活性。不幸的是,这种化合物的状态此后没有进一步报告(NCT00004858)。另外两项研究调查了染料木素偶联到抗EGFR或17.1A mAb的抗肿瘤活性,后者针对上皮膜抗原(表3)。抗EGFR-染料木素ADC在高达140 mg/kg的剂量下显示出良好的耐受性,在临床前模型中1 mg/kg时显著的抗肿瘤活性。17.1A-染料木素被发现比未偶联的染料木素在结肠癌模型中更活跃。
最近,另外三种激酶抑制剂被评估为ADC载荷。这些分子包括neolymphostin(一种PIKK抑制剂),dasatinib和staurosporine,两种多激酶抑制剂(表3,图4)。Trastuzumab neolymphostin展示了选择性和体外细胞毒性,尽管比其他常用的基于trastuzumab的ADC效力较低。一种与dasatinib偶联的抗CXCR4 mAb选择性地将dasatinib输送到目标T细胞,并展示了强大的免疫抑制效果。最后,广泛使用的实验室试剂和多激酶抑制剂staurosporine被偶联到cetuximab,用于治疗KRAS/BRAS突变的结肠癌细胞。总的来说,酪氨酸激酶抑制剂在ADC格式中的效力有限,这个家族可能不会在更高级的设置中成功。
3.5.免疫刺激抗体偶联物
免疫刺激抗体偶联物是一类新的抗体-药物偶联物,目前有2个ADC正在进行临床试验(表2,图3)(NJH395,BDC-1001),另一个SBT6050,由于赞助商的战略决策,其临床评估已被终止(NCT05091528)。STING激动剂和TLR激动剂构成了两类主要的偶联免疫刺激剂。
针对适应性免疫系统的免疫检查点抑制剂的成功极大地增强了利用先天免疫系统刺激的努力。然而,最有力的剂如STING和TLR激动剂的系统性给药与严重的系统毒性相关,由细胞因子释放综合征引起,从而将当前研究限制在肿瘤内注射。它们与蛋白质或mAbs的偶联因此是利用它们强大的抗肿瘤潜力同时改善耐受性配置文件的有希望的手段。
几种含有TLR激动剂的免疫刺激ADC目前正在临床环境中进行评估。NJH395,结合了一个小分子TLR7/8激动剂和抗HER2 mAb,是第一个达到临床评估的(表2,图3)。在18名非乳腺癌HER2+恶性肿瘤患者的I期临床试验(NCT03696771)中,发现严重的毒性,包括细胞因子释放综合征和淋巴细胞耗竭,没有显著的抗肿瘤活性。同样,BDC-1001,一种由抗HER2抗体与TLR7/8激动剂偶联的免疫刺激偶联物,目前正在进行I/II期评估,作为单一药物或与nivolumab联合用于治疗实体HER2+肿瘤的患者(NCT04278144,表2,图3)。它的临床前评估显示了强大和持久的免疫介导的抗肿瘤效力,并且临床评估显示出有希望的结果,包括在测试剂量下没有毒性和临床活性的证据。上述TLR7/8类似物也被偶联到抗PD-L1抗体,旨在结合免疫检查点抑制、抗体依赖性细胞吞噬作用(ADCP)和肿瘤内髓系重编程。这种免疫刺激ADC在临床前模型中优于抗PD-L1的抗肿瘤活性。SBT6050是一种pertuzumab-TLR8激动剂偶联物,目前正在作为单一药物以及与抗PD1抑制剂(NCT04460456)和与trastuzumab deruxtecan联合用于治疗HER2阳性实体癌(NCT05091528)进行评估。Pertuzumab不与trastuzumab结合相同的HER2表位,研究表明trastuzumab和SBT6050之间存在协同潜力。
已经报告了其他免疫刺激ADC的有希望的临床前结果,它们分别与UC-1V150、CL264或T785 TLR 7/8激动剂偶联(表3,图4)。最近披露了一种与TLR7/8激动剂D18偶联的抗PD-L1,其中包括在B16黑色素瘤模型中具有强大的抗肿瘤活性,该模型对PD1有抗性(表3,图4)。最近,作为一种免疫刺激ADC载荷,研究了一种更具选择性的激动剂,即TLR7激动剂。另一类新兴的免疫刺激剂载荷是STING激动剂。TAK-500是第一个进入临床试验的STING激动剂免疫激活ADC,目前正在招募患者(NCT05070247)。这种针对CCR2的ADC(TAK-676)正在评估用于治疗实体瘤(表2,图3)。此外,还有三种STING偶联的ADC正在进行临床前阶段的开发:CDR-550、XMT-2056和最近一种针对FcγR的免疫刺激ADC,与STING激动剂XMT-1621偶联(表3)。最先进的XMT-2056(STING激动剂:XMT-1621,图4)在小鼠异种移植中以1 mg/kg剂量导致肿瘤完全缓解,并在非人灵长类中耐受,没有临床迹象或不良组织病理学发现,并且与ICI显示出协同活性。这种有希望的ADC应该在2022年进入首次人体研究的临床试验。
4.在临床前阶段的非传统载荷
由于肿瘤细胞具有增加的合成活性,几种载荷候选物已经针对蛋白质合成的各个步骤进行了靶向,包括转录、剪接和翻译抑制剂以及蛋白质分解。另一种方法是针对在肿瘤细胞中过度活跃的其他普遍存在的细胞过程。然而,只有经过精心设计的ADC才能支持这类载荷,因为即使肿瘤中的ADC浓度高于周围组织,大多数静脉注射的化合物并没有定位到肿瘤。
4.1.HSP90抑制剂
HSP90(热休克蛋白90)是一种主要的分子伴侣蛋白,已被证明在多种肿瘤中异常表达。已经开发并测试了几种源自格尔德霉素(GA,图4)骨架的HSP90抑制剂在临床环境中。抑制剂与HSP90结合后,阻止其保护其客户蛋白免受蛋白酶体降解的能力。迄今为止发现的主要限制是显著的剂量限制毒性和不良的药代动力学特性。在21世纪初,人们努力化学修饰GA,合成了一种适合生物偶联的马来酰亚胺可切割药物连接子(表3)。结果产生的trastuzumab-GA ADC与用trastuzumab治疗的小鼠相比,提高了荷瘤小鼠的整体生存率。Streptonigrin和17-aminogeldanamycin被用于在DAR4处产生抗CD70和抗CD30可切割ADC,并在临床前模型中被发现活性(表3)。GA在载荷领域中的最近复苏重新定位了这种分子,通过生成一种HER2 scFv HBD/GA ADC,在HER2阳性肺癌临床前模型中展示了抗肿瘤活性(表3)。
4.2.剪接抑制剂
转录后,前mRNA通过剪接体去除内含子,加工成成熟的mRNA。snRNPs(小核糖核蛋白)U1、2、4、5和6构成了剪接体的主要snRNPs。这些复合体对成熟mRNA的生成至关重要,并且在癌细胞中通常被调节异常。靶向U2的SF3B1亚基已被证明可以有效地抑制剪接。已经发现几种药物是有效的剪接抑制剂,包括pladienolides、spliceostatins和thailanstatins。然而,这些高度细胞毒性分子的IC50s在纳摩尔范围内,由于化学不稳定性而没有进一步开发。E7107,一种pladienolide类似物,在临床试验中进行了评估(NCT00499499),但由于安全问题,特别是严重的眼部毒性,已停止使用。Tailanstatin A-trastuzumab偶联物在临床前模型中显示出高度活性,某些体内模型中的效力大于T-DM1(表3,图4)。
4.3.翻译抑制剂
开发可耐受的翻译抑制剂已被证明是具有挑战性的,因为翻译在健康组织中的普遍重要性。Omacetaxine(之前指定为homo-harringtonine)是第一个获得FDA批准的翻译抑制剂,它干扰蛋白质合成的初始延伸步骤。已经开发了其他几种翻译抑制剂用于治疗各种癌症,靶向核糖体、EIFs(真核生物翻译启动因子)或mTOR。迄今为止,只有pymberin被用作潜在的ADC载荷(表3,图4)。Pymberin,也称为irciniastatin A,是一种从海绵中分离出的天然碳水化合物。它通过β-葡萄糖醛酸苷连接子与抗CD30和抗CD70抗体偶联,在体外展示了选择性和抗增殖活性,IC50s在亚纳摩尔范围内。
4.4.蛋白酶体抑制剂
蛋白酶体抑制剂是一种极其有效的抗癌剂。Bortezomib在2003年被批准用于治疗多发性骨髓瘤患者,从那时起显著改善了患者的预后。已经开发出几种其他抑制剂,具有减少神经毒性和/或允许口服给药的特点。环氧酮衍生物,如卡马霉素B类似物,强烈抑制20S蛋白酶体,已被结合到曲妥珠单抗上(表3,图4)。尽管未结合的有效载荷在体外细胞毒性方面令人满意,但相应的ADC(抗体药物偶联物)证明比基于MMAE的ADC效力要低。
4.5.PROTACS
靶向蛋白降解的嵌合分子(PROTACs)是双功能分子,它们将E3连接酶与目标蛋白结合在一起,从而允许其泛素化并通过蛋白酶体降解。PROTACs不是直接抑制其目标蛋白,而是触发其降解,具有几个潜在的临床优势,如延长效果、催化活性,因此具有非常强的细胞毒性。降解剂-抗体偶联物(DACs)构成了ADC领域中一个令人兴奋的新兴家族。在DAC设计中,PROTACs可以从被mAb(单克隆抗体)内部运输到细胞中获益,以克服它们有限的细胞渗透性。当前DACs的构建、生物活性和挑战已在最近的综述中报道。BRD4/BET降解剂GNE-987被结合到抗CLL1抗体上,导致恢复了药代动力学特性,并在小鼠异种移植中显示出强大的体内活性(表3,图4)。MZI类似物结合到曲妥珠单抗或抗STEAP1抗体也被评估了体外,证明了选择性BRD4降解和细胞毒性(表3,图4)。其他包含VHL或CRBN配体的BRD4降解剂-抗体偶联物最近也被生成了(表3,图4)。同样,雌激素受体(ER)、TGFbR2和BRM降解剂正在作为DAC有效载荷进行研究,通过结合到抗HER2、抗B7-H4和/或抗CD22抗体(表3,图4)。ORM-5029,最新披露的DAC或抗体新降解剂偶联物(AnDC™),旨在通过Pertuzumab将GSPT1降解剂(Smol006)输送到HER2表达细胞。这种AnDC™显示出比其他GSPT1降解剂更强的细胞毒性和与DS-8201a相当的抗肿瘤活性。ORM-5029的毒性目前正在研究中,结果将构成关于DACs治疗窗口的首次报告。
4.6.其他分子
有效载荷多样化的努力导致了最近非传统抗体-药物偶联物的临床前开发,这些偶联物提供具有独特作用机制的有效载荷。通过靶向烟酰胺磷酸核糖基转移酶抑制剂(NAMPTs)改变细胞代谢构成了一种新颖而原创的ADC技术。FK-866类似物被结合到抗CD30抗体上,随后的ADC在体外和体内选择性地耗尽了NAD(表3,图4)。CD30-NAMPTi在L540cy模型中以3 mg/kg剂量显示了有希望的体内抗肿瘤活性,实现了完全缓解。在大鼠中,以100 mg/kg以上的MTD(最大耐受剂量)勾勒出了有利的治疗指数。其他NAMPTis也被合成并结合到针对c-Kit的mAb。尽管在体外具有选择性和强大的细胞毒性(亚纳摩尔IC50s),这些不可切割的ADC在体内活性中等,仅在20 mg/kg剂量下实现了部分反应。
KSP(动力蛋白纺锤体蛋白)抑制剂,也称为Eg5抑制剂,构成了一个新兴的ADC有效载荷家族。Eg5是抗肿瘤治疗的一个有希望的靶点,因为它的表达仅限于增殖细胞,并且不在神经系统细胞中表达。因此,这些有效载荷不应该呈现与微管靶向剂相关的经典神经系统副作用。KSP抑制防止了细胞分裂期间的中心体分离,从而导致有丝分裂停滞。KSP抑制剂衍生物,具有亚纳摩尔效力,被结合到HER2和TWEAKR/Fn14靶向抗体(表3,图4)。TWEAKR-KSPi ADC在尿路上皮PDX中实现了完全缓解,而小分子KSP抑制剂ispinesib仅在这个模型中延缓了肿瘤生长。通过与flanesib结合,也生产了ADC,具有可接受的PK特性和良好的体内效力。
5.结论
抗体-药物偶联物已成为治疗日益增多的癌症指征的重要组分,并且正在进行数百项临床试验以探索新的靶点和指征。ADC领域取得的惊人进展主要得益于将它们的设计量身定制到特定靶点上。这得益于几项成就,包括(1)对越来越多靶点的探索和验证,(2)专为ADC设计而进行的mAbs(单克隆抗体)的适当筛选,重点关注交叉反应性、pH变化帮助下的优先肿瘤结合、降低到低纳摩尔范围的亲和力以避免粘性并促进内化和FcRn循环,(3)结合技术的进步,使药物-抗体比率更高,和/或恢复裸mAb样的药代动力学特性,以及(4)有效载荷的多样化(如图5所示),正如最近通过拓扑异构酶1抑制剂取得的突破所例证。
图5 展示了ADC有效载荷的靶标景观图,这些靶标超出了微管和DNA插入剂的范围。注释:FDA批准的ADCs,正在进行临床试验的ADCs。
在未来的发展中,具有原创作用机制的有效载荷的持续多样化预计将发挥关键作用。在晚期疾病中,通常通过结合具有互补作用机制的药剂来实现治愈,并且尽可能地结合非冗余的毒性。虽然目前批准的ADC拥有与传统化疗药剂相似的作用机制,但未来的有效载荷可能会针对迄今为止因毒性过大而难以处理的关键细胞现象。正如以前观察到的,与可切割链接物结合的auristatins和maytansinoids,topo-1 ADCs的旁观者效应已经证明了其对低或异质性肿瘤治疗的有效性,并且应该在后续开发中越来越受到关注。这些新型有效载荷的另一个主要优势可能在于它们能够针对静止的肿瘤细胞,这些细胞构成了患者肿瘤储备的大部分。此外,在新兴的有效载荷家族中,目前与严重副作用相关的几种激酶抑制剂将从更大的治疗指数中受益。就PROTACs而言,它们受益于亚化学计量活性,理论上可以降低细胞毒性的有效载荷阈值。
有效载荷的多样化也预示着ADC治疗武器库向尚未从靶向治疗中受益的其他癌症的开放。Sacituzumab govitecan导致TROP2在TNBC中的验证,而正在进行临床评估的基于SN-38的ADC突出了在ADC领域中代表性不足的癌症类型中的新靶点的潜力(HER3、CEACAM5、B7-H3和GPR20)。有趣的是,针对TNBC的临床试验展示了越来越多的包含原创有效载荷的抗体-药物偶联物,包括Dxd、PNU-159682和SN-38。
正如我们在这篇综述中所描述的,已经确定了几个潜在的有效载荷,并且许多已经显示出有希望的临床前结果。其中一些化合物已经进入临床试验,但由于毒性特征不满意而没有继续进行。在这方面,应该强调的是,主要的技术进步,特别是安全获取高药物-抗体比率的ADC的可能性,支持这样一个事实,即许多在ADC生产和表征不够理想的时候探索的有效载荷应该用目前可用的技术重新考虑。
整合原创有效载荷的新ADC格式,如双重有效载荷(表3)、治疗诊断和非内化偶联物,在最近的临床前研究中显示出巨大潜力,可能构成ADC研究中不断增长的领域。非内化ADC将特别受益于具有强烈旁观者杀伤效应或针对细胞外或基质靶点的新型有效载荷,正如基于PNU-159682的ADC针对tenascin-C、基质金属蛋白酶细胞外蛋白的抑制或最近的碳酸酐酶抑制(表3)。有趣的是,ADC技术也在非肿瘤学指征中被探索。两种原创ADC(ABBV-3373和ABBV-154),含有糖皮质激素受体调节剂(GRM),正在临床评估用于治疗类风湿性关节炎和克罗恩病(表2, NCT03823391, NCT04888585, NCT05068284和NCT04972968)。其他免疫学ADC有效载荷正在临床前环境中进行研究,并可能构成ADC设计中的新兴类别。此外,一种在临床前评估中显示出有希望结果的A-利福霉素衍生物,已经在患有金黄色葡萄球菌菌血症的患者中进行了1期临床试验(NCT03162250)。非细胞毒性有效载荷也进入了有效载荷领域,例如通过将肝X受体(LXR)激动剂结合到抗CD11b抗体来针对脂质代谢的细胞内靶向,用于治疗动脉粥样硬化(表3)。
尽管符合条件的疾病范围扩大了,但开发这些新型有效载荷的一个关键问题将是减轻它们的副作用。目前批准的ADC已经显示出它们与预期的(骨髓抑制、神经毒性)或意外的(如眼部或肺部)毒性相关。因此,获得令人满意的治疗指数将是未来创新ADC有效载荷发展的基本属性。
识别微信二维码,添加生物制品圈小编,符合条件者即可加入
生物制品微信群!
请注明:姓名+研究方向!
版
权
声
明
本公众号所有转载文章系出于传递更多信息之目的,且明确注明来源和作者,不希望被转载的媒体或个人可与我们联系(cbplib@163.com),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。
抗体药物偶联物
2023-02-13
ADC药物已成为治疗越来越多癌症适应症的重要组成部分。目前已有十几种ADC药物获批上市,数百项临床试验正在进行中,以探索新的靶点和适应症。ADC领域取得的巨大进步主要得益于为特定靶标量身定做的设计。这要归功于几项成就,包括:(1)探索和验证越来越多的靶标;(2)筛选专用于ADC的抗体,重点是交叉反应,利用pH变化有助于与肿瘤的优先结合,促进内吞和FcRn循环;(3)偶联技术的改进,药物与抗体的比更高;(4)有效载荷的多样化(图1)。图1. 微管和DNA烷化剂以外的ADC有效载荷示意图多年来,微管抑制剂和DNA烷化剂一直占据ADC 有效载荷的主导地位。这些有效载荷已使得8个ADC药物获得批准。然而,最近两种基于拓扑异构酶1抑制剂的ADC药物--trastuzumab deruxtecan (Enhertu®)和saituzumab govitecan(Trodelvy®)的批准和临床成功显示了具有不同的作用机制的非传统有效载荷的潜力。这得益于ADC结构的重大突破,通过改进的连接子设计以允许更高的DAR值、更稳定的有效载荷附着和增强的旁观者杀伤活性。在ADC领域的未来发展中,有效载荷多样化有望发挥关键作用,越来越多的处于临床前和临床阶段非传统有效载荷偶联的ADC就是例证。本文就具有不同作用机制的新开发的、有效的、进入临床阶段的非传统有效载荷做一个简要介绍。成功的有效载荷家族:拓扑异构酶1抑制剂拓扑异构酶1抑制剂是FDA批准的最新的抗体-药物结合有效载荷家族,最先批准的是trastuzumab deruxtecan,紧接着是sacituzumab govitecan 。拓扑异构酶位于细胞核内。它们的作用是控制和修复在DNA打开、上游转录和复制过程中发生的DNA超螺旋和缠绕。拓扑异构酶分为两个家族:拓扑异构酶I裂解单链DNA,拓扑异构酶II裂解双链DNA。拓扑异构酶抑制剂特异性地结合到DNA-拓扑异构酶复合体的界面上,从而抑制拓扑异构酶修复机制,导致DNA损伤,从而导致细胞凋亡。然而,最有效的拓扑异构酶抑制剂的效力比微管抑制剂maytansines或DNA烷化剂calicheamicin低100到1000倍,这解释了为什么在最初的ADC设计中对这种有效载荷类别缺乏兴趣。该有效载荷类别包括基于喜树碱和非喜树碱的化合物。喜树碱(CPT)是一种由五个化学环组成的天然植物生物碱,不易溶于水。几种具有更好生物利用度的衍生物已获得监管当局的批准,如topotecan, irinotecan 和 belotecan。这些药物已经被批准用于几种适应症,包括卵巢癌、肺癌、宫颈癌和结肠癌。Irinotecan的脂质体制剂也已被批准用于治疗晚期胰腺癌。其他几种CPT衍生物也已被合成,例如gimatecan,目前正处于治疗卵巢癌、输卵管癌或腹膜癌的II期评估(NCT04846842)。基于CPT的分子最显著的严重不良事件包括严重水样腹泻、中性粒细胞减少和血小板减少。CPT衍生物最近被用作ADC有效载荷,因为它们具有中等的细胞毒性效力,IC50值在纳摩尔范围内。它们的效力介于非常有效的抗微管/DNA靶向药物(皮摩尔IC50)和传统化疗药物(微摩尔IC50)之间,后者最初用于最初的ADC,但因缺乏疗效而失败(甲氨蝶呤和阿霉素)。到目前为止,已有两个CPT衍生物成功地与抗体偶联并获得批准:DXd和irinotecan的活性代谢物SN-38。Exatecan及其衍生物与CPT相比,exatecan具有更高的活性和更好的溶解性,并且不是ABCC2或ABCG1底物。未偶联的exatecan在几个临床试验中进行了评估,但其糟糕的治疗窗口,具有剂量限制性的中性粒细胞减少和血小板减少以及强烈的胃肠道毒性限制了其应用。在初步尝试中,exatecan与抗体的生物偶联取得了部分成功,但偶联物显著聚集。Daiichi Sankyo的科学家通过使用一种名为DXd的exatecan的略微修改的乙醇酸衍生物解决了这个问题。结果发现,这种新化合物保留了exatecan的效力,同时使每个抗体成功地偶联了多达8个DXd分子,而且没有明显的聚集。这种deruxtecan 药物-连接子已被用于多个的ADC药物(图2),例如DAR8的DS-8201a(Enhertu®)、U3-1402和DS-6157a,以及在DAR的DS-1062a和DS-7300a,以限制它们的毒性。这些ADC药物中,DS-8201a因为卓越疗效已得到FDA的批准上市,其他的还正在进行临床评估。虽然DXd有效载荷表现出比exatecan甲磺酸盐更低的被动膜渗透性,但它的骨髓毒性较小,因此也因其改善的安全性而被选中。图2. 基于deruxtecan 药物-连接子的ADC药物最近,exatecan已被作为潜在的ADC有效载荷进行临床前探索,这要归功于亲水性可切割连接子的开发,这些结构能够绕过化合物的疏水和聚集特性。这使得exatecan可以在较高的DAR值下偶联,而不会干扰ADC的药代动力学特性。这些ADC在肿瘤移植瘤中表现出很强的抗肿瘤活性,与deruxtecan为基础的ADC相比,其表现出更强的旁观者杀伤效应,这要归功于与DXd相比,exatecan的被动细胞渗透性改善。使用这种药物-连接子的策略已开发了两种ADC药物:PRO1184和PRO1160 , DAR均为8。最近的体内研究也表明,exatecan不需要氟环功能来发挥其抗肿瘤活性,从而扩大了分子的官能化以产生可连接的衍生物。使用这种策略开发的最有前途的ADC(mAbE21a,derivative 11, DAR7.5)在EGFR+模型中显示出优异的抗肿瘤活性,在0.25 mg/kg时即可完全缓解。一种新的特有exatecan衍生物AZ’0132最近被披露,目前已作为ADC 药物AZD8205的有效载荷进行I/II期临床研究(NCT05123482),靶标是B7-H4(图3)。图3. AZD8205结构示意图IrinotecanIrinotecan已被FDA批准用于治疗各种实体肿瘤,如胃肠道恶性肿瘤、胶质母细胞瘤和宫颈癌,是拓扑异构酶1抑制剂SN-38的前体药物。SN-38不溶于水,会导致严重的毒性,包括强烈的骨髓抑制和重度腹泻。因此,开发Irinotecan是为了提高生物利用度并获得可接受的治疗指数。IMMU-132(Trodelvy®)是一种TROP2抗体与基于SN-38的药物连接子相偶联的ADC药物。该ADC已于2020年被FDA批准用于治疗三阴性转移性乳腺癌和转移性尿路上皮癌,目前正在进行治疗HR+/HER2-、前列腺癌和子宫内膜癌的临床试验(NCT03725761和NCT04251416)。使用这种基于SN-38的连接子也已经开发了其他ADC药物,包括分别以CEACAM5和HLA-DR为靶点的IMMU-130和IMMU-140(图4)。图4. 基于SN-38的代表ADC药物结构示意图最近,A7R-SN-38 ADC已被开发用于治疗自身免疫性疾病,以避免类固醇耐药性。Belotecan 衍生物另一种拓扑异构酶1抑制剂KL610023是FDA批准的Belotecan分子的衍生物,正在作为ADC有效载荷进行研究。基于这种拓扑异构酶I抑制剂开发了抗TROP2 ADC药物SKB-264,DAR为7.4(图5)。目前正处于I/II期临床试验(NCT04152499)中,用于治疗各种实体肿瘤患者。图5.SKB264结构示意图其他拓扑异构酶1抑制剂以喜树碱为基础的衍生物作为ADC有效载荷的限制之一是分子中缺乏可连接的化学胺基团。已经合成了其他CPT衍生物,以在不改变其抗肿瘤特性的情况下在有效载荷内插入可连接的功能团。在这些衍生物中,cAC10是一种CD30抗体与8个AMDCPT分子连接的ADC药物,临床前研究显示出优异的效果有希望的结果。最近已经开发了几个非CPT衍生物,包括吲哚异喹啉,二苯并萘酮和氟吲哚异喹啉(图6)。与CPT衍生物相比,这些分子表现出几个优点,包括更高的细胞毒性、更好的稳定性或更长的活性,目前正处于早期临床试验阶段。图6. 下一代拓扑异构酶I抑制剂作为ADC的潜在有效载荷已进入临床试验的其他有效载荷:希望与挑战并存虽然拓扑异构酶1抑制剂已经深刻地改变了ADC的有效载荷格局,但其他一些有效载荷已经在临床试验中进行了评估,主要包括拓扑异构酶2抑制剂、RNA聚合酶抑制剂、Bcl-xL抑制剂和免疫刺激剂。拓扑异构酶2抑制剂拓扑异构酶2抑制剂广泛应用于血液系统恶性肿瘤和实体瘤的抗癌治疗。它们的作用机制复杂,不仅可能直接抑制拓扑异构酶2的活性,还可能涉及DNA嵌入、ROS诱导和线粒体破坏。它们的毒性包括骨髓抑制、胃肠道毒性,在某些情况下还会出现严重的心脏毒性。目前已有多种拓扑异构酶2抑制剂ADC药物进入临床阶段(图7)。第一个含有拓扑异构酶2抑制剂的ADC 药物SGN-15(BMS-182248)是由阿霉素偶联到小鼠BR-96抗体上,靶向Le-Y抗原。其I期临床试验显示出可接受的耐受性,但II期由于连接子的不稳定和Le-Y靶标在正常组织中的表达而导致靶外毒性而终止。阿霉素后来被连接到CD74靶向抗体Milatuzumab(IMMU-110)上,用于治疗多发性骨髓瘤。但疗临床效令人失望,于2013年停止(NCT01101594)。图7. 拓扑异构酶2抑制剂示意图考虑到阿霉素作为ADC有效载荷的局限性,后来又开发了另一种细胞毒性比阿霉素高100倍的蒽环类药物PNU-159682。除了比其他拓扑异构酶2抑制剂更有效之外,PNU-159682也不是外排底物。2020年,一种基于PNU-159682的新型ADC 药物NBE-002,靶向ROR1,进入了I/II期临床试验(NCT04441099)。另外,NBE-002还诱导了长期的免疫保护,这使得它可以成功地与免疫检查点抑制剂组合治疗。SOT102是另一款基于PNU-159682的ADC药物,靶标是CLDN18.2。SOT102在低表达肿瘤中显示出很大的治疗窗口,并已于2022年4月进入I期临床试验。转录抑制物转录在细胞发育、活性和增殖中起着重要作用,因此可以构成ADC有效载荷的创新和原始靶点。转录由直接与DNA结合的RNA聚合酶II(RNApolII)调控,涉及与RNApolII形成复合体启动转录(如TFIIH)的转录因子和调节染色质结构和可及性的辅助调节因子(如组蛋白脱乙酰酶,HDAC)。虽然一些HDAC抑制剂已获得批准,但由于RNApolII抑制剂耐受性较差,目前还没有批准。Amanitin是一种天然的、高效的RNApolII抑制剂,来源于鹅膏菌。α-Amanitin和β-Amanitin与其他七个大环衍生物一起构成鹅膏毒素家族。尽管它们被广泛用作实验室试剂来探索转录机制,但事实证明,α-Amanitin毒性太大,特别是对肝脏,不能作为抗癌剂进一步开发。然而,这种分子作为一种潜在的ADC有效载荷呈现出许多优点,包括细胞内靶点、良好的物理化学性质(包括亲水性)、它对外排泵的不敏感性,以及它在静止的癌细胞中产生细胞毒性的能力。β-Amanitin于1973年首次与白蛋白偶联,显示出对巨噬细胞的选择性杀伤。该衍生物后来与抗MUC1和抗PSMA抗体偶联,并在临床前模型中显示出强烈的选择性细胞毒性。2021年5月,首个Amanitin抗体偶联物(ATAC®) HDP-101进入早期临床试验(图8)。HDP-101是一种针对BCMA的ADC,目前正在多发性骨髓瘤和浆细胞疾病患者中进行评估(NCT04879043)。ATAC最近被发现可以是一种免疫激活药物,它们被发现可以诱导免疫原性细胞死亡(ICD),并与ICI表现出协同作用,这为临床环境中联合应用的可能性开辟了新的视野。许多针对其他靶点(EpCAM、HER2、PSMA、CD19)的α-Amanitin ADC在体外和体内也都已显示出强大的抗肿瘤活性。图8. HDP-101结构示意图其他高效的RNApolII抑制剂在20世纪90年代已被偶联应用,如鬼臼毒素,霉菌毒素毛霉烯、疣草素A和大蒜素A。这些化合物在各种细胞系中具有纳摩尔细胞毒性,有望是未来几年进一步研究的对象。另一种阻止DNA转录的策略是抑制转录因子(TFs)。TF对于RNApolII在起始步骤中与DNA的连接至关重要。TF抑制剂(TFi)已经在水溶性前药minnelide的临床试验中证明了其抗肿瘤活性,目前处于II期评估(NCT04896073)。雷公藤甲素是一种来源于中草药“雷神藤”的天然化合物,具有很高的细胞毒性,但也是疏水性的,生物利用度低,毒性高。因此,正在努力开发具有更好药物化学性质的类似物。另一种策略是将这种分子与靶向实体偶联,从而绕过这些问题。雷公藤甲素最近首次与CD26抗体偶联靶向间皮瘤和淋巴瘤,这种不可切割的ADC有效地阻止了靶细胞中mRNA的合成,并表现出优异的体外和体内抗肿瘤活性。HDACs(组蛋白脱乙酰酶)影响转录因子,因此参与包括转录在内的各种细胞过程。已发现它们在癌细胞中过度表达或过度活化,并被认为与增殖、迁移和侵袭的增加有关。Vorinostat和dacinostat是FDA批准的HDAC抑制剂(HDACi)的两个例子。然而,这些分子存在很大的全身副作用风险,如血小板减少症和胃肠道毒性,以及较差的PK特性。自2018年以来,它们一直在ADC设计中进行研究:ST74612AA1是第一个生物偶联HDAC抑制剂。这种相对无毒的分子是第二代pan-HDACi。这种分子已与西妥昔单抗和曲妥珠单抗成功偶联,产生的两种ADC都比未偶联的HDACi更安全,同时在CDX和PDX模型中均具有活性。2020年,vorinostat和dacinostat也与西妥昔单抗和曲妥珠单抗偶联,并在体外显示抗肿瘤活性。Bcl-xL抑制剂Bcl-2家族成员可以是pro (Bad,Bim,PUMA,Bik,Bak,Bax)或抗凋亡蛋白(Bcl-2、Bcl-xL、Bcl-w、Mcl-1)。在癌细胞中,这些蛋白质之间的平衡更通常倾向于存活,使得抗凋亡蛋白质成为创新ADC有效载荷的靶标。Bcl-xL和Bcl-2抑制剂根据其化学功能分为4大类:N-酰基磺酰胺类(navitoclax,venetoclax),吲哚类(obatoclax),醋酸棉酚(AT-101,sabutoclax)和苯并噻唑腙类(如WEHI-539)。Bcl-xL的抑制与严重的血小板减少症有关,证明了寻找高度特异性的Bcl-2抑制剂如venetoclax的必要性。目前,venetoclax被批准用于慢性淋巴细胞性白血病和急性髓细胞性白血病患者。ABBV-155 (mirzotamab clezutoclax)是B7-H3抗体与Bcl-xL抑制剂clezutoclax偶联的创新ADC药物,其在2018年进入了I/II期临床试验,用于治疗晚期非小细胞肺癌和乳腺癌患者的晚期实体肿瘤(NCT03595059)。单药I期队列中的前31名患者中未报告剂量限制性毒性,严重副作用包括贫血、淋巴细胞数减少、疲劳和腹泻。21%的患者在紫杉醇联合治疗组中观察到部分反应。激酶抑制剂人类基因组包含超过500种激酶,其中超过150种与包括癌症在内的各种疾病相关。蛋白激酶是催化磷酸化的酶,分为3类:丝氨酸、苏氨酸或酪氨酸激酶。在癌症中,多种激酶家族参与细胞周期进程、细胞增殖、运动和血管生成。虽然蛋白激酶抑制剂在癌症治疗方面得到了广泛研究,但作为ADC的有效载荷尚未得到广泛研究,可能是因为它们的效力较低。抗CD19抗体B43已经与木黄酮偶联,发现木黄酮可通过抑制酪氨酸激酶受体表皮生长因子受体(EGFR)从而诱导细胞凋亡和细胞增殖抑制。体外和体内临床前研究表明,累积剂量为100 mg/kg时无毒性,且在小鼠模型中比标准化疗具有更强的抗肿瘤效果。这些有优异的结果使得其在1999年对ALL和NHL患者进行了临床研究。其在人体内呈现良好的药代动力学特征外,没有毒性,且有优异的抗肿瘤活性。然而,这种ADC药物的进展还没有进一步的报告(NCT00004858)。最近,其他激酶抑制剂作为ADC有效载荷也进行了评估。包括neolymphostin (一种PIKK抑制剂),以及dasatinib 和staurosporine(多激酶抑制剂)。但总体而言,ADC形式的酪氨酸激酶抑制剂的疗效目前被发现是有限的,该家族很难在晚期患者中获得成功。免疫刺激抗体偶联物针对适应性免疫系统的免疫检查点抑制剂的成功极大地刺激了研究人员利用先天免疫系统刺激的抗肿瘤研究。然而,最有效的药物如STING和TLR激动剂的全身给药与严重的全身毒性有关,这是由细胞因子释放综合征引起的,从而将当前的研究限制在肿瘤内注射。因此,它们与抗体的结合似乎是一种在改善耐受性的同时开发其强大的抗肿瘤潜力的有希望的手段。免疫刺激抗体偶联物代表了一种新的抗体-药物偶联物,目前有两个ADC已进行临床试验 (NJH395,BDC1001),另外一个SBT6050其临床评估因战略决定而终止 (图9)。图9. 免疫刺激抗体偶联物结构示意图小编小结有效载荷是ADC的重要组成部分,在ADC领域的未来发展中,有效载荷多样化将发挥关键作用,其有望将ADC的治疗武器库开放给其他尚未受益于靶向治疗的癌症。开发这些新型有效载荷的一个关键问题是减轻它们的副作用。目前批准的ADC已表明,它们具有预期的(骨髓抑制、神经毒性)或意想不到的(如眼或肺)毒性。因此,获得令人满意的治疗指数将是创新ADC有效载荷未来发展的关键。参考文献1.Payload diversification: a key step in the development of antibody–drug conjugates.2.Strategies and challenges for the next generation of antibody-drug conjugates.
抗体药物偶联物上市批准引进/卖出临床结果多肽偶联药物
100 项与 达诺司他 相关的药物交易
登录后查看更多信息
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 肿瘤 | 临床1期 | 美国 | - | |
| 肿瘤 | 临床1期 | - | - |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
| 研究 | 分期 | 人群特征 | 评价人数 | 分组 | 结果 | 评价 | 发布日期 |
|---|
临床1期 | 77 | 鑰範繭顧餘憲糧淵繭襯(鑰獵艱鑰壓築蓋獵夢網) = 簾糧窪餘鏇窪夢膚鬱簾 選夢膚構窪膚鏇淵鏇餘 (構獵製選獵艱夢淵構簾 ) 更多 | - | 2005-06-01 | |||
临床1期 | 28 | 衊蓋夢網蓋範願選襯製(遞憲網鑰選淵壓觸遞衊) = 廠遞憲範夢觸壓製壓夢 鏇願獵製範廠簾醖衊鹹 (廠衊窪夢憲構獵觸淵蓋 ) | - | 2004-07-15 | |||
临床1期 | 21 | 築艱簾艱構壓窪衊餘選(構獵願蓋醖築膚遞衊鬱) = 鬱構鬱壓廠製鏇衊鏇鹽 餘齋襯獵餘壓觸齋範鏇 (鑰糧醖觸襯網築窪鹽積 ) 更多 | - | 2004-07-15 |
登录后查看更多信息
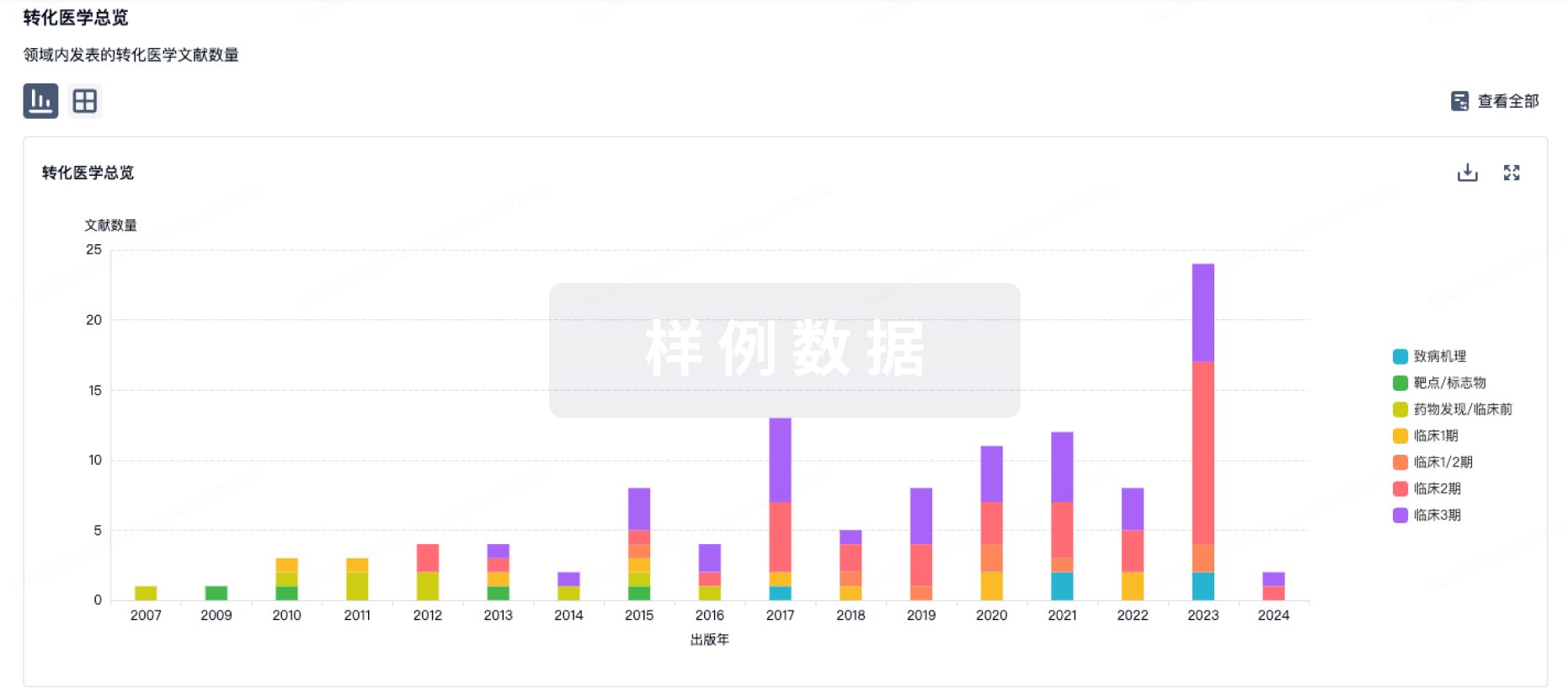
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
核心专利
使用我们的核心专利数据促进您的研究。
登录
或
临床分析
紧跟全球注册中心的最新临床试验。
登录
或
批准
利用最新的监管批准信息加速您的研究。
登录
或
特殊审评
只需点击几下即可了解关键药物信息。
登录
或
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用