更新于:2024-05-01
Intercept Health
更新于:2024-05-01
概览
关联
100 项与 Intercept Health 相关的临床结果
登录后查看更多信息
0 项与 Intercept Health 相关的专利(医药)
登录后查看更多信息
81
项与 Intercept Health 相关的新闻(医药)2024-01-16
非酒精性脂肪肝炎(Nonalcoholic Steatohepatitis,NASH)是一种由非酒精性脂肪肝(Nonalcoholic Fatty Liver Disease,NAFLD)进展而来的严重慢性肝脏疾病。根据流行病学调查,全球的NAFLD患病率约为25%,其中10% - 30%最终会发展为NASH,这意味着全球的NASH患者在1.9亿至5.8亿之间[1]。
2020年,NASH全球药物市场增涨至19亿美元,并有望在2025年突破百亿大关,开发NASH药物的市场前景极为可观。虽然NASH药物需求的蛋糕越来越大,然而,目前为止尚未有NASH适应症的药物批准上市。
2018年维生素E和吡格列酮获得FDA优效评价,但中国临床诊疗指南并未推荐该用药方案,原因是其在日本中心的试验结果不理想,且药物长期用药有中毒风险。其他临床应用有一定时间的药物,他汀类和贝特类降脂药、二甲双胍等药物对NASH的疗效也均未在《中国非酒精性脂肪肝病防治指南》中得到认可[2]。适用脂代谢异常的药物用于NASH治疗还需要进一步的上市后临床试验验证。
真正可改善肝代谢、改善炎症和逆转纤维化的NASH治疗药物仍在被全球市场期待。本文将梳理NASH药物的研发进展。
NASH治疗药物作用机制可以分为四类:改善糖脂代谢异常、缓解肝脏炎症、降低脂肪毒性减少肝细胞死亡以及抑制或改善肝纤维化,对应NASH的病理状态[1]。值得注意的是,药物的作用机制并不是单一的,同一种药物可以通过多种作用机制治疗或缓解NASH,如FXR激动剂除了能改糖脂代谢异常状态,还具有抗纤维化的作用。
NASH的真正病因是体内代谢失衡,因此针对糖脂代谢开发的靶点及对应进入临床试验的药物最多,这种倾向从全球著名药企NASH在研管线布局中即可瞥见。但从NASH病理的复杂性可看出,未来的NASH药物处方可能类似其他营养代谢疾病,倾向于多机制调节药物或多种药物联合用药。
临床试验情况
针对NASH开展的临床试验大多集中在临床I期(65例)和II期(155例),临床III期的开展数量相对较少,仅20例。靶点布局方面FXR激动剂、GLP1类似物在研发热度上远超其他靶点。
值得注意的是,闯入临床III期决赛圈的药物,多在其他适应症中已成功获批上市。例如在美国被临床诊疗指南推荐有限度用于NASH病人的FXR激动剂[3]奥贝胆酸上市适应症为胆汁性肝硬化;诺和诺德的明星药GLP-1类似物semaglutide已获批二型糖尿病和肥胖症;同样的还有糖尿病药PPARγ激动剂吡格列酮和SGLT2抑制剂恩格列净等,实际上均为已上市药物,重新就NASH适应症开展临床试验。该部分药物由于安全性已被充分验证,因此即使对NASH的疗效不佳,部分也在临床诊疗指南中被酌情推荐使用。
真正以NASH为主要适应症进入临床III期的未上市药物只有Madrigal的THRβ激动剂Resmetirom,该药原研公司为Roche。其他针对NASH开发新药、新靶点大多数还处在早期的临床概念验证或药效验证阶段,有效性还等待临床试验进一步讨论
临床阶段药物的靶点前10位集中针对改善糖脂代谢异常,FXR、GLP-1R、PPAR为临床试验申报数最多的药物靶点。
药物类型分布
不追热点,另辟蹊径是否又能摘得桂冠呢?生物制药、siRNA技术或许为NASH治疗药物带来新的可能,另外通过调节肠道菌群纠正人体正常糖脂代谢的策略也被考虑用于NASH治疗。
生物制药产品拥有药代清晰、非靶效应少的优势,小RNA药物能够靶向一些难以成药的靶点,肠道菌调节治疗代谢疾病这一概念也在今年来被广泛认可。
生物药以GLP-1R和FGFs类似物居多,另外Cytodyn的CCR5单抗Leronlimab进入临床II期,Merck和Ngm Biopharmaceuticals合作的GDF15类似物NGM395进入临床I期。
小RNA药物方面,针对HSD17B13的RNA干扰药物包括Aligos Therapeutics与GSK合作开发的ARO-HSD和Alnylam Pharmaceuticals同类药物ALN-HSD进入临床II期;百时美施贵宝和Nitto Denko Corporation靶向HSP47的小RNA也进入临床II期;Aligos Therapeutics与Merck今年1月也宣布会加大合作开发适用于NASH的siRNA药物。
肠道菌疗法方面,慕恩(广州)生物科技有限公司开发的胃肠道微生物调节剂MNO-163处在临床前阶段;LISCure Biosciences的细菌替代物LBP6在临床I期;Mikrobiomik Healthcare开发的MBK-01进入临床II期,均探索肠道菌群调节糖脂代谢的可能性。
生物制药、siRNA技术与肠道菌调节技术打破不可成药靶点的铠甲,在NASH药物开发路程上另辟蹊径。
优效或积极评价药物
截止目前为止共有24项NASH相关临床试验获得优效或积极评价,试验集中在II期临床。在获得优效或积极评价的药物看见了更多新靶点的面孔。受试药物的靶点仍然集中在糖脂代谢调控。
开发企业中,辉瑞凭借果糖激酶抑制剂PF-06835919、DGAT1/2抑制剂Ervogastat和ACC抑制剂Clesacosta 3款药物拔得头筹,ervogstat和clesacostat的组合疗法在2022年5月更是活得FAD授予针对伴纤维化的NASH适应症快速通道资格;诺华凭借SGLT1/2双重抑制剂Licogliflozin和DGAT1抑制剂Pradigastat 2款药物紧随其后。
Conatus开发的一款caspase抑制剂Emricasan通过降低脂毒性保护肝脏细胞,成为众多优效受试药物中唯一不针对代谢调节机制的药物,只可惜虽然获得了积极评价,但最终试验结果仍然没有显示药物有改善患者NASH和先前存在肝纤维化的患者的肝脏炎症或纤维化的能力。
值得注意的是这些试验中目前只有3例继续往前推动,且除了已上市药物奥贝胆碱和二甲双胍外,继续挺入临床III期的只有Inventiva的泛PPAR激动剂Lanifibranor,该药物已获得FDA突破性疗法称号,今年1月刊登在《柳叶刀》子刊上的文章明确Lanifibranor对改善肥胖病人NASH评分的效果,是NASH药物开发赛道上的一名热门选手[4],国内正大天晴已正式引进该药。
除了Lanifibranor外,2022年还有不少NASH新靶点治疗药物临床研究的好消息:
今年1月89bio的FGF21类似物pegozafermin在一项1b/2a期概念验证临床试验中获得积极结果。
2月Axcella Therapeutics新药AXA1125获得FDA授予的快速通道资格,适应症为NASH伴肝纤维化;AXA1125是一种代谢调节剂,目前已进入临床II期。
5月Gilead的ACC1、ACC2双重抑制剂宣布了II期临床试验的积极结果。9月,Akero Therapeutics的FGF21类似物efruxifermin在2b期临床试验中也取得积极数据。
真正以NASH为适应症开发的药物陆续取得捷报,未来NASH用药有望突破老药新用的局限,呈现靶点及药物百花齐放的局面。
根据药渡数据,处于IND申报阶段之前,靶点明确的NASH药物共85例(除却无进展及终止项目)。前10位布局的靶点基本与进入临床III期的靶点一致。
结合临床试验进展,不难看出FXR激动剂、GLP-1R激动剂和PPAR的赛道十分火热。学术界广泛认可这3类靶点治疗机制,《柳叶刀》子刊今年发表的1篇文献认可了这3类靶点对NASH的治疗作用[4]。
然而,众多企业对热门靶点布局,也注定在这些管线中终将有一大部分将面临失败或失去市场空间。开辟新药物类型,如抗体药、siRNA药物靶向难以成药靶点或自主开发新靶点或许才是后起之秀们反超的唯一办法。
FXR激动剂
FXR,法尼酯x受体,激活FXR可以抑制胆固醇7α羟化酶,从而减少胆汁酸的生成;上调肝细胞上的胆盐输出泵,从而增强胆汁酸从肝细胞向胆小管转运;下调肝脏牛磺酸钠协同转运体,增加胆汁酸在肝中的的重吸收;上调回肠胆汁酸结合蛋白,增加胆汁酸在肠中的重吸收。以上各种调控最终使得脂肪吸收量减少。
此外激活该受体可以降低血浆甘油三酯,提升HDL水平。Intercept开发的奥贝胆酸是最有望在众多FXR抑制剂中突出重围获得NASH适应症批准的药物。虽然临床瘙痒副作用一直为人诟病,同时国内临床指南并不承认其对NASH的疗效,2020年奥贝胆碱申请NASH适应症上市更是被FDA拒绝,但今年Intercept宣布奥贝胆酸III期临床试验终于达到试验主要终点,再次申报获批的概率大大增加。
推荐项目:项目合作|FXR激动剂
FGFs类似物
FGF21、FGF19由胆汁酸与FXR结合后经肠道内释放。修饰后的FGF类似物不具有刺激增殖和致癌性质,而保留了其对脂肪代谢的调控作用。比起FGFs类似物具有胰岛素增敏和抗肝脏纤维化的作用,对肝硬化也有效果。
89bio的FGF21类似物pegozafermin在临床1b/2a期结果中获得积极评价,诺和诺德的FGFs调节剂NN-9499目前已进入临床II期,国内东阳光生物药研发有限公司开发的HEC-88473也正在开展临床I期试验。
GLP-1R激动剂
胰高血糖素样肽GLP-1,可激动GLP-1R有促进胰高血糖素降解,抑制胰高血糖素分泌,增加胰岛素的分泌,提高机体胰岛素敏感性的作用,是目前常用的抗糖尿病药。
最近研究表明,GLP-1类似物具有减重效果,已获FDA批准肥胖症、超重等适应症用于减肥。肥胖会导致糖脂代谢异常,加重NASH病情,因此GLP-1R激动剂也是潜在的NASH治疗靶点。然而诺和诺德的GLP-1类似物semaglutide今年也宣布其针对NASH开展的临床II期试验没有达到理想终点。但这并不意味着GLP-1R激动剂的策略彻底失败。
糖脂代谢是一个复杂的过程,接受着众多激素的调控,Altimmune、Zealand Pharma和道尔生物研发的GLP-1R/GIPR双重激动剂,或者多种NASH靶点药物联用都有可能让GLP-1R激动剂突破重围成为首获批NASH适应症的药物。
PPAR(α、γ、δ)激动剂
PPAR(α、γ、δ)是一类过氧化氢酶体增殖物激活受体。激活PPARα可以促进脂肪酸氧化和促进脂肪细胞分化。激活它可以增强脂蛋白脂酶的活性加速脂蛋白的分解,同时也能减少肝脏中脂蛋白的合成,从而降低血脂(降低甘油三酯,提升HDL)。贝特类降脂药,为其临床使用的激活剂,主要通过纠正血脂异常来发挥抗动脉粥样硬化作用。
PPARγ正常情况下由游离脂肪酸激活并促进胰岛素分泌。吡格列酮就是常用的PPARγ激动剂,虽然它对该靶点的激活作用较弱,但是其对mTOT也有一定作用,使得其在NASH可能中有疗效,目前已进入III期临床,试验其联合维生素E治疗NASH的效果。然而使用该类药物有体重增加、增加骨折风险和体液潴留的副作用。
然而在国内临床诊疗中对PPARα和γ的治疗效果有所保留,并未推荐用于NASH治疗。PPARδ在巨噬细胞和kupffer细胞中均有表达,激活它可以抑制炎症。但是单纯激活该靶点也会引起能量代谢异常,导致类似胰岛素抵抗的代谢综合征出现。
由于单独激动某一亚型的PPAR受体均有局限性,目前NASH的药物开发策略是开发泛PPAR多亚型激动剂,Inventiva的PPAR三重激动剂Lanifibranor在临床II期获得有效,有望完成III期试验顺利上市;国内的杭州百诚医药科技股份有限公司布局PPARα和δ双重激动剂;深圳微芯生物科技股份有限公司原研PPAR三重激动剂西格列他。
然而,Genfit的PPARα和δ双重激动剂Elafibranor在III期试验中期没有达到试验预期,它的失败给PPAR激动剂对NASH的开发蒙上了不确定性,但也能为PPAR激动剂临床试验方案提供经验。
THRβ激动剂
THRβ,甲状腺素受体β,它的激活除了产生甲状腺素的相关功能,还被证明具有降低脂毒性物过载的作用,并能够调节血脂,使肝脏快速“去脂”。
Madrigal Pharmaceuticals开发的THRβ激动剂Resmetirom已进入临床III期,2019年《柳叶刀》杂志已发表其II期临床试验结果,充分肯定其有效性,不良反应为中轻度可耐受[5]。
Viking Therapeutics的VK2809在II期临床取得出色结果,目前多国II期试验在同步开展,试验有望在2023年12月结束。
ACC1、ACC2抑制剂
ACC,乙酰辅酶A羧化酶。参与脂肪酸的重头合成过程,抑制脂肪的氧化。抑制它的活性可以减少脂肪酸的生成。有研究表明它的抑制会引起SREBP-1代偿性激活,阻碍脂肪酸进一步生成甘油三酯,减轻高甘油三酯血症。
Gilead Sciences的ACC1、ACC2双重抑制剂GS-0976管线进度到临床II期并于今年5月获得积极结果;辉瑞的Clesacostat单用已完成II期临床试验,与DGAT1、DGAT2双重拮抗剂Ervogastat联用的II期试验更是得到优效评价并获得FDA快速通道资格。这些成功弥补了辉瑞PF-05175157和默沙东MK-4074失败的阴霾,重新让ACC抑制剂走上NASH治疗的赛道。
CCR2、CCR5拮抗剂
CCR2-CCR5信号轴,可以放大肝脏内的非特异性免疫响应,并引发炎症,从而使肝星形细胞激活,肝脏发生纤维化。拮抗信号轴被认为是逆转、改善纤维化,降低炎症防疫的手段。Tobira和Takeda原研的CCR2-CCR5双重拮抗剂Cenicriviroc曾在临床II期试验中失败,然而数据再分析显示出其抗纤维化作用,并被艾尔建收购继续开展III期临床试验,目前试验已终止,但具体结果分析尚未公布。Cenicriviroc和诺华FXR激动剂Tropifexor联用方案也在II期试验中。国内广东众生药业和药明康德也共同布局了CCR2、CCR5拮抗剂WXSH-0213。
DGAT1、DGAT2
DGAT-1、DGAT-2是一种甘油酰基转移酶,主要作用机制是使二酰甘油加上脂肪酸酰基形成三酰甘油,参与脂肪代谢,脂类在组织中扮演重要角色。辉瑞的DGAT1、DGAT2双重抑制剂Ervogastat,Ionis Pharmaceuticals的DGAT2抑制剂IONIS-DGAT2Rx均已走上临床II期。青岛黄海制药的DGAT-1抑制剂WXFL-50010210已获得中国I类新药称号,目前处在临床I期。
SUMMARY
小结
NASH作为严重慢性代谢性疾病,其病因复杂,但最终带来的结果是肝脏糖脂代谢紊乱,肝纤维化及肝功能受损,引发肝硬化甚至诱发肝癌。NASH目前的药物需求市场巨大,然而尚未有获批NASH适应症的药物,我国临床诊疗指南并不承认常用的糖尿病、高血脂药物对NASH的治疗效果。
所幸NASH的药物研发火热,处于临床III期、II期药物种类和靶点类型数目众多,联用方案也已开展临床试验,不少药物获得FDA快速通道资格,相信在不久的将来NASH适应症药物将会陆续上市。面对NASH复杂的病理状态,未来很有可能迎来多药或多靶点治疗NASH的用药局面。
2024-01-13
·药事纵横
声明:因水平有限,错误不可避免,或有些信息非最及时,欢迎留言指出。本文仅作医疗健康相关药物介绍,非治疗方案推荐(若涉及);本文不构成任何投资建议。据不完全统计,过去的2023年里有31款新药被FDA拒批,涉及到肿瘤、心脏疾病、呼吸系统疾病和皮肤病等等,被拒理由绝大多数集中于对产品有效性和安全性的质疑上,包括临床获益存疑、临床数据单一等问题。除临床问题外,CMC(化学、制造和控制)问题也开始成为FDA拒绝批准的另一项重要因素。在此文中小编整理了2023年FDA拒批新药,或许能为业内理解FDA的审评标准提供帮助。一月:礼来2023年1月19日,礼来发布公告表示公司已经收到FDA关于其阿尔茨海默病新药donanemab加速批准申请的完整回复函。FDA没有批准的原因是NDA申请所基于的临床试验中接受至少12个月药物治疗的患者数量有限。FDA特别要求礼来提供至少100名接受donanemab至少12个月持续治疗的患者的数据。FDA在完整回复函中未提及其它缺陷。礼来公司在发布会上解释说:“虽然该试验包括100多名接受donanemab治疗的患者,但由于斑块减少的速度,许多患者能够早在治疗6个月就停止给药,导致不到100名患者接受了12个月的donanemab治疗。”二月:Soligenix、Cytokinetics2月14日,Soligenix宣布收到FDA的RTF(Refuse to file)信函,意味着FDA否决了SGX301用于皮肤T细胞淋巴瘤(CTCL)的新药申请(NDA)。经过初步审查,FDA认为该公司于2022年12月14日递交的NDA不完整,无法进行实质性审查。受此消息影响,Soligenix公司股价暴跌约40%。2月28日,Cytokinetics宣布Omecamtiv mecarbil的上市申请收到FDA的完全回复函(CRL)。Omecamtiv mecarbil是一种新型的选择性小分子心肌肌球蛋白激活剂,可增强心肌收缩力,且不增加心肌细胞内的钙浓度或心肌耗氧量。FDA认为三期临床GALACTIC-HF的数据没有建立充分的有效性证据,不足以支持其获批上市。FDA要求进行一项额外的临床试验来建立Omecamtiv mecarbil的有效性证据。三月:Veru、Incyte、艾伯维3月4日,FDA拒绝了Veru公司sabizabulin药物的紧急使用授权(EUA)申请,该药物用于治疗患有中度至重度Covid-19的住院成人患者,这些患者患有ARDS(急性呼吸窘迫综合征)的风险很高。Veru首席执行官Mitchell Steiner表示,FDA拒绝使用该药是因为可能存在未知影响,或研究的不确定性。在咨询委员会之前准备的简报文件中,FDA对Veru提交的关键数据表达了担忧。FDA表示,在小型试验中,虽然sabizabulin达到了降低死亡率的目标,但指出了不确定性,即使这些不确定性在个体上没有问题,但对结果提出了疑问。3月22日,艾伯维宣布,已收到FDA就ABBV-951(foslevodopa / foscarbidopa)新药上市申请发出的完整回复函。在信中,FDA要求艾伯维提供ABBV-951设备(泵)的额外信息作为NDA的一部分,不需要再额外做与该药相关的疗效和安全性试验。艾伯维表示,将会尽快重新提交NDA。3月23日,Incyte宣布,FDA已就JAK1/JAK2抑制剂芦可替尼缓释片(每日1次)的上市申请发出了完整回复函(CRL)。该产品拟用于治疗某些类型的骨髓纤维化、真性红细胞增多症和移植物抗宿主病。FDA承认,Incyte所提交的研究达到了基于曲线下面积(AUC)参数的生物等效性目标,但还提出了额外的批准要求。Incyte计划与FDA会面以确定下一步行动。四月:礼来、诺和诺德4月13日,礼来宣布,收到美国FDA针对公司提交的mirikizumab治疗溃疡性结肠炎(UC)的生物许可证申请(BLA)发出完整的回复函,回复函中,FDA列举了mirikizumab生产相关问题,对该药物的临床数据、安全性或药品说明书未提出问题。 礼来表示,他们正在与FDA积极沟通合作,希望尽快在美国市场推出mirikizumab药物。但礼来没有给出解决制造问题可能需要多长时间的答案。诺和诺德在2023Q1财报中透露,已于4月24日收到FDA就concizumab用于预防治疗伴抑制物的A型和B型血友病的生物制品许可申请(BLA)发出的完整回复函(CRL)。FDA要求其提供额外的与患者相关的监测和给药信息,以确保concizumab按预定方案给药。此外,诺和诺德还需要提交额外的concizumab生产过程材料文件。五月:ImmunityBio、Byondis5月1日,Ascendis Pharma宣布收到FDA就TransCon PTH用于治疗成人甲状旁腺功能减退症(HP)的新药申请(NDA)发出的完整回复函(CRL)。基于该结果,Ascendis计划与FDA召开A类会议以确定TransCon PTH的后续上市计划。Ascendis表示,FDA在CRL中对TransCon PTH药品或设备无法保证均一的递送剂量表示担忧,但并未对其临床数据提出异议也未要求该公司开展新的临床前研究或临床研究。5月11日,ImmunityBio表示,该公司IL-15超级激动剂Anktiva(N-803)与卡介苗联合治疗膀胱癌的上市申请被美国FDA拒绝批准。此次拒批主要是由于FDA在对其第三方合同制造商进行许可前检查时发现了缺陷。FDA还就如何解决问题给出了建议,指出了ImmunityBio必须解决的制造问题,还要求其在重新提交的文件中更新Anktiva组合的某些安全性和有效性数据。5月15日,Byondis公司宣布收到FDA的完整回复函(CRL)。FDA在CRL中表示,暂停对靶向HER2的ADC药物SYD985生物制品许可申请(BLA)的批准,需要更多的信息来支持审批决定,这些信息也需要额外的时间来审查。Byondis方对FDA的决定感到遗憾,但是也表示将继续推进SYD985在欧盟和英国的上市。六月:F2G、Aldeyra、Intercept、再生元6月19日,F2G公司宣布收到FDA就olorofim用于治疗侵袭性真菌感染的上市申请发出的完整回复函(CRL)。FDA在CRL中称,该公司需要提交额外的临床数据及相关分析。该公司仍对Olorofim表示乐观,并将满足FDA的要求并继续寻求批准。6月21日,生物技术公司Aldeyra Therapeutics, Inc.宣布收到FDA对候选药物ADX-2191治疗原发性玻璃体视网膜淋巴瘤新药申请(NDA)的完全回应函。虽然没有发现ADX-2191的安全或生产问题,但FDA表示,由于基于文献的NDA提交中 "缺乏充分和良好的对照调查",因此 "缺乏实质性的有效性证据"。近日,公司宣布ADX-2191治疗视网膜色素变性患者的2期临床试验积极顶线结果。相对于基线,临床试验证明了在许多不同的生理和心理物理评估中视网膜功能的统计学显著改善。6月22日下午,Intercept宣布FDA已经对该公司用于治疗非酒精性脂肪性肝炎(NASH)引起的肝硬化前纤维化的奥贝胆酸(OCA)新药申请发出了完全回应函(CRL)。FDA在CRL中表示,经审查确定,奥贝胆酸不能以目前的形式获得批准。根据CRL的内容,在NASH中重新提交OCA的NDA至少需要成功完成REGENERATE研究的长期结果阶段。这次受挫后,Intercept决定停止所有与NASH相关的投资,重组公司业务以加强其对罕见和严重肝脏疾病的关注,并从2024年开始加速实现盈利。6月27日,再生元(纳斯达克:REGN)宣布,已收到FDA就阿柏西普8mg制剂的上市申请发出的完整回复函(CRL)。公司表示,FDA此次拒绝批准是因为在第三方药物灌装机中发现了生产问题。与疗效、安全性、说明书和原料药生产等方面无关,也未要求提供额外的临床数据或额外开展试验。阿柏西普药物用于治疗湿性年龄相关性黄斑变性(wAMD)、糖尿病性黄斑水肿(DME)和糖尿病性视网膜病变(DR) 。再生元表示,将与FDA和第三方供应商密切合作,尽快将阿柏西普8 mg制剂带给wAMD、DME和DR患者。七月:Amneal、Citius Pharmaceuticals7月3日,生物制药公司Amneal Pharmaceuticals(NYSE: Amneal)表示,FDA拒绝批准公司用于治疗帕金森病的IPX203新药申请(NDA),完整回应函(CRL)中表示是因为该药物的安全性数据不足,无法帮助患者长期控制症状。与IPX203的功效或生产问题无关。此外,FDA表示,虽然该公司根据研究确定了一种成分左旋多巴的安全性,但它无法充分确定另一种成分卡比多巴的安全性。FDA已要求提供更多信息。此次NDA的提交是基于RISE-PD 3期临床试验的结果。7月29日,Citius Pharmaceuticals公司公布收到FDA就LYMPHIR™(用于治疗持续性或复发性皮肤T细胞淋巴瘤患者)的BLA申请发出的完整回复函(CRL)。Citius Pharmaceuticals公司表示,FDA要求补充强化产品检测,并增加控制措施。同时与BLA一起提交的安全性和有效性临床数据包或拟议的处方信息没有问题。八月:Mesoblast、渤健/Sage、Galera、Outlook8月4日,Mesoblast宣布Remestemcel-L治疗儿科激素耐药的急性移植物抗宿主病(SR-GVHD)的上市申请收到DA的完全回复函(CRL),FDA要求更多数据以支持上市批准。Mesoblast将进行一项针对死亡率更高的高危成人患者的对照试验。FDA希望该公司提交更多的数据来支持BLA的批准,并希望其在开展新的临床试验之前先解决remestemcel-L的化学、生产和质量控制(CMC)问题。8月7日,Sage Therapeutics公布Q2业绩,并宣布因FDA拒绝了与渤健合作的Zuranolone用于治疗重度抑郁症(MDD)的上市申请。FDA发布了关于zuranolone治疗MDD的NDA的完整回复函(CRL)。CRL指出,公司对于MDD的新药申请中,没有提供实质性的有效性证据来支持zuranolone治疗MDD的批准。如想寻求在MDD上的批准,Sage/Biogen至少需要对该药物进行一项额外的研究。Sage和Biogen正在审查反馈,并评估接下来的步骤。8月9日,Galera Therapeutics宣布收到FDA就Avasopasem manganese(Avasopasem,GC4419)用于治疗接受标准治疗的头颈癌(HNC)患者因放疗引起的严重口腔粘膜炎(SOM)的新药上市申请(NDA)发出的完整回复函(CRL)。FDA在CRL中表示,III期ROMAN研究以及IIb期GT-201研究数据不足以证明Avasopasem在减少头颈癌患者严重口腔黏膜炎方面的有效性与安全性。FDA认为,需要重新递交额外的临床试验结果。8月30日,Outlook Therapeutics宣布收到FDA就ONS-5010(贝伐珠单抗)用于治疗湿性年龄相关性黄斑变性(wAMD)的上市申请发出的完整回复函(CRL)。FDA在CRL中表示,虽然ONS-5010的关键III期NORSE TWO研究达到了有效性和安全性的主要终点,但是由于一些CMC(化学、生产和质量控制)问题、批准前生产检查的开放观察以及缺乏实质性证据,无法在当前的审查时间内批准其上市。ONS-5010的上市申请主要基于3项III期临床试验(NORSE ONE、NORSE TWO和NORSE THREE)的积极结果。九月:阿斯利康9月6日,阿斯利康宣布FDA拒绝了其长效C5补体抑制剂Ultomiris的补充申请,该申请寻求批准该药物用于治疗抗水通道蛋白4(AQP4)抗体阳性(Ab+)的成年视神经脊髓炎谱系障碍(NMOSD)患者。FDA发布了一份关于Ultomiris补充生物制品许可申请的完整回应函(CRL),CRL中并未对Ultomiris的疗效和安全数据提出意见,仅要求修改Ultomiris风险评估和缓解策略(REMS),并对患者的脑膜炎球菌疫苗接种状况进行背景调查,或要求在治疗前预防性使用抗生素。阿斯利康将通过其罕见病部门 Alexion与 FDA 密切合作,以确定如何最好地调整REMS 计划。Ultomiris新适应症NMOSD距离在美国上市又近了一步。十月:礼来、Alnylam、Alvotech、再生元/赛诺菲10月2日,美国FDA拒绝批准礼来lebrikizumab治疗中度至重度特应性皮炎(湿疹)的生物制剂许可申请(BLA),原因是在一家第三方制造商中发现了问题。来自美国监管机构的完整回应函(CRL)中没有提到对lebrikizumab的临床数据、安全性或标签的担忧,这增加了延迟不会太久的希望——假设CMO的问题可以解决或生产转移到新的合作伙伴。礼来公司表示,没有其他药物受到CRL的影响,将“继续与第三方制造商和FDA密切合作,以解决反馈意见,使患者能够获得lebrikizumab”。10月9日,Alnylam宣布其收到了FDA就RNAi疗法Patisiran(商品名:Onpattro)补充上市申请(sNDA)发出的完整回复函(CRL)。根据CRL的内容,FDA认为Alnylam提供的现有临床数据不足以支持Patisiran用于治疗转甲状腺素蛋白淀粉样变性心肌病(ATTR-CM)的上市申请。鉴于此结果,Alnylam将不再寻求扩大Patisiran在美国的适应症范围,而是将重点转向另一款治疗ATTR-CM的RNAi疗法——vutrisiran的III期研究。2023年10月12日,Alvotech公司宣布美国食品和药物管理局(FDA)拒绝批准其针对AVT04(乌司奴单抗)的生物制剂许可申请(BLA),该公司提议将AVT04作为强生公司Stelara(ustekinumab)的生物仿制药候选药物,这是该公司自去年以来第四次被监管机构拒绝批准。监管机构在其完整回复函(CRL)中指出,Alvotech位于冰岛雷克雅未克的生产设施存在“缺陷”,FDA在2023年3月的现场检查中发现了这些缺陷。除此之外,FDA没有指出其AVT04的BLA中的任何其他问题,该公司打算近期重新提交BLA。这将触发另一个为期六个月的审查周期和新的生物仿制药用户费用修正案(BsUFA)目标行动日期。10月20日,再生元和赛诺菲宣布收到美国FDA就Dupixent(度普利尤单抗)治疗慢性自发性荨麻疹(CSU)的补充生物制剂许可申请(sBLA)发出的完整回复函(CRL)。CRL指出,需要额外的疗效数据来支持批准。再生元表示一项正在进行的临床试验(Study C)将继续招募患者,预计在2024年底获得结果,将提供额外的疗效数据。十一月:Aldeyra11月27日,Aldeyra Therapeutics宣布收到了美国食品和药物管理局(FDA)针对治疗干眼症的候选新药reproxalap的新药申请(NDA)发出的完整答复函。虽然没有发现 reproxalap 的安全性或生产问题,但FDA在信中指出,该NDA没有证明对治疗干眼症相关眼部症状的疗效,至少应再进行一项充分且对照良好的研究,以证明对治疗干眼症眼部症状的积极作用。根据NDA审查周期的剩余时间,FDA可能无法在《处方药用户费法》(PDUFA)的目标行动日期2023年11月23日或该日期前后批准该 NDA,并要求Aldeyra开展额外的临床试验并提交这些临床试验的结果,然后才会重新审议该申请。十二月:Checkpoint 、默沙东、Zealand、安进12月18日,Checkpoint Therapeutics宣布其抗PD-L1抗体Cosibelimab用于治疗转移性或局部晚期皮肤鳞状细胞癌(cSCC)的生物制剂许可申请(BLA)因生产问题被FDA拒批。FDA已就cosibelimab BLA发布了一份完整答复函(CRL)。CRL仅仅指出了在对公司的第三方合同生产组织进行多赞助商检查时发现的问题,作为重新提交批准申请时需要解决的问题。CRL没有对cosibelimab 的批准性的临床数据包、安全性或标签表示任何担忧。12月20日,默沙东宣布FDA已对其非麻醉性、口服选择性P2X3受体拮抗剂gefapixant用于治疗成人难治性慢性咳嗽(RCC)或不明原因慢性咳嗽(UCC)的新药上市申请申请(NDA)发出完整回复函(CRL)。FDA认为,默沙东的申请没有足够的证据证明gefapixant能有效治疗RCC和UCC。CRL与gefapixant的安全性无关。默沙东表示正在审查FDA的反馈,以确定下一步的措施。8、Aldeyra:reproxalap治疗干眼症。2023年12月23日,Zealand Pharma宣布,美国食品和药物管理局(FDA)在发现第三方生产基地存在问题后,拒绝了Zealand的胰高血糖素类似物dasiglucagon用于治疗先天性高胰岛素血症(CHI)。CRL并未对dasiglucagon的临床数据包或安全性提出任何担忧。Zealand公司计划于2024年上半年重新提交NDA的第一部分,前提是对相关地点的重新检查取得成功。该公司还希望根据FDA的要求,对其在CHI的NDA第二部分中有关dasiglucagon使用超过三周的连续血糖监测数据集进行补充分析。2023年12月26日,安进宣布,FDA已完成对Lumakras(sotorasib)新药补充申请的审查,拒绝完全批准Lumakras用于治疗既往至少接受一次全身治疗的KRAS G12C突变非小细胞肺癌患者。FDA要求安进开展额外的验证性研究,并不晚于2028年2月完成。总结美国一直是创新药的超级市场,被FDA批准上市往往相当于拿到了全球通行证。但近年来FDA在创新药的上市审批上日渐趋严,以高标准、高要求将多款创新药被拒之门外。上市申请被拒除了影响产品的商业化进程外,对于严重依赖于某一单品的小型制药公司更可能是致命的,很多公司在公布CRL消息后都遭遇了重大的股市打击。参考资料:各公司公告
临床3期加速审批
2024-01-11
·研发客
// 2023年多款引发热议的新药被FDA拒之门外,如KRAS抑制剂sotorasib、RNAi疗法patisiran、NASH新药奥贝胆酸以及干眼症新药reproxalap。被拒绝的原因不一,以临床有效性不足和CMC问题为主。同时有多款同领域新药首次获得FDA批准,例如首个治疗杜氏肌营养不良(DMD)的基因药物Elevidys、首个CRISPR基因编辑疗法Casgevy。更有意思的是,同是渐冻症治疗药物,渤健的tofersen能获得FDA青睐,而另两个新药CNM-Au8和NurOwn则未获得认可。拒绝或批准一款创新药,FDA审评的考量因素纷繁复杂。研发客梳理了2023年FDA审评的部分新药案例。可观察到的是,FDA的审评始终贯穿着满足患者临床需求的科学理念,综合评估药品风险获益比,而不是简单的缩紧和放松。临床疗效证据不足据统计,FDA在2023年共发出32封完整回复函(CRL, complete response letter)拒绝创新药批准,其中有21个新药因临床疗效不足被拒绝。例如RNAi疗法patisiran,虽3期临床达到研究终点,但FDA认为产品治疗转甲状腺素蛋白淀粉样变性心肌病(ATTR-CM)没有明显的治疗效果,临床证据不足。相反,部分新药虽未达到临床终点,仍获得FDA批准。如基因疗法Elevidys在2023年6月获得FDA加速批准治疗杜氏肌营养不良(DMD),但支持产品BLA申请的临床研究错过了主要功能终点。带来的问题是,FDA批准和拒绝一款新药时的审评逻辑如何?前FDA资深临床审评员、现思路迪医药首席医学官肖申博士认为,FDA对于新药审评并不完全是非黑即白,不是简单依据临床有效性终点是否达到,还要参考安全性结果评估,最主要的是考虑患者需求,从而根据最终的风险获益分析来决定药物是否获批。“如果该适应症领域无药可治,FDA考虑临床急需的迫切性,会适当放宽要求,如DMD新药Elevidys、治疗渐冻症的tofersen。如果该领域已有其他药物获批,FDA标准会相对严格,获批标准的门坎会进一步提高,要求后面的药物有确切的疗效和/或更高的安全性。”肖申说。例如ATTR-CM适应症,在patisiran被拒前,另一个新药tafamidis已先行获批相同适应症,且临床疗效更优,可帮助患者延长寿命并减少因心血管疾病住院次数。相比下,patisiran的临床3期研究虽统计学上也达到了主要终点,但并未真正确切反映其临床价值,只是轻微改善患者的体能状况和生活质量。类似的案例还有KRAS抑制剂sotorasib,针对KRAS突变肺癌已有其他治疗药物,且sotorasib的确证性临床研究未能证明疗效有突破性优异之处。“患者需求永远是第一位的,”肖申表示,“唯一的不确定因素,是如果研究未达到主要临床终点,但达到次要终点,这类情况需要进行综合评估,以及咨询外部专家顾问委员会的建议。”再以治疗杜氏肌营养不良的Elevidys为例,最终以生物标志物——微肌营养不良蛋白为替代终点获得批准,早期也以8:6投票的微弱优势通过专家委员会讨论。“虽然Elevidys没有达到主要终点,但次要终点有明显获益,病人从地板起身的时间、10米步行或跑步时间的相对变化都有改善,且药物相对也比较安全,从患者需求、产品疗效及安全性的综合评估,最终FDA批准了该药。”肖申表示。“整体来说,FDA的新药审评是一项系统工程,需要整体风险获益评估。例如有些药物尽管有很多风险,但疗效很好,且真正满足病人所需,因而获批。同时上市后的监管也同样重要,如黑框警示和REMS(风险评估和缓解策略)的应用等。FDA一年多前批准一个治疗肥厚性心肌病的药物,要求所有处方该药物的医生必须经过严格训练、获得证书后,才能给病人处方该药。”肖申概括道。替代终点未获认可事实上,在2023年FDA拒绝的多个新药中,很多产品变更临床主要终点,或使用替代终点以获取研究成功,但仍未获得FDA认可,例如渤健/Sage的zuranolone、Intercept的奥贝胆酸,以及Aldeyra公司的reproxalap。肖申认为,替代终点的选择须建立在同时具备质和量的基础上,能够真正预测有临床意义变化的基础上。通常情况下,替代终点选择会参考指南推荐,若指南没有推荐,可以和FDA沟通选择替代终点,但FDA即使同意了该替代终点,也要提供足够的临床数据,达到一定量化指标,以确认替代终点能够真正反映药物的疗效。以治疗非酒精脂肪性肝炎(NASH)的奥贝胆酸为例,其3期关键研究REGENERATE使用的替代终点是肝纤维化程度改善和NASH症状消除,也是FDA指南认可的临床终点。已公布的中期数据中,奥贝胆酸25mg组在18个月时,22.4%的患者实现纤维化改善≥1期且NASH没有恶化,安慰剂组是9.6%,另一终点NASH症状消除未显示益处。最终FDA专家委员会以12:2投票反对药物批准,认为试验使用的替代终点获益不明显,对2或3期NASH纤维化患者的风险大于获益。FDA也在CRL中表示,要提供患者的长期随访数据才能重新申请NDA。FDA对奥贝胆酸的审评结果也印证了肖申的观点,即替代终点选择必须要提供充分量化数据,可靠预测临床获益。近期,nefecon(布地奈德缓释胶囊)获得FDA完全批准,是首个以蛋白尿作为替代终点获得加速批准的IgA肾病药物。作为当年该药的FDA临床审评员,肖申表示,在2016年之前,FDA并不接受蛋白尿是IgA 肾病确定的有效替代终点。但FDA心肾审评部联合美国肾脏病协会、美国国家肾脏病基金会、以及数家大学和制药公司的多次合作,通过大量回顾性研究,最终证实蛋白尿与预测IgA肾病疗效的相关性,加速批准该药上市。而对于其他慢性肾病,蛋白尿的作用并未确定。除了考虑量的变化,药物作用机制也是衡量替代终点获得批准的基础。例如肌萎缩侧索硬化(ALS)治疗药物tofersen,以生物标志物神经丝轻链(NfL)作为替代终点,于2023年4月获批上市。同是治疗ALS,FDA却拒绝了纳米疗法CNM-Au8以NfL减少的研究结果获得加速批准。另一个同样被拒的干细胞ALS治疗药物NurOwn,其NfL改善数据也未被认可。除患者需求、以及首个批准的因素外,肖申认为,不同药物的作用机理不同,比如对于ALS的新药开发,有些药物的NfL改变可以预测临床结果、改善患者获益,而有些则不行。虽是同一个生物标志物,但预测的结果不同,须明确药物作用机理,才能进一步预测该生物标志物是否能预测临床获益。“总的来说,替代终点本身可能仅是某个实验室指标或影像学指标,将其用于判断临床疗效挑战性很大,选择合适的替代终点是一项非常复杂的工程,需要企业和监管机构甚至学术界共同合作研究确定。”肖申表示。CMC要求更严格除临床问题外,CMC(化学、制造和控制)问题也开始成为FDA拒绝批准的另一项重要因素。2023年FDA发出的32封CRL中,与CMC相关共有11项。相比临床研究受到挑战,大部分因CMC被拒企业的态度是积极的,且部分产品在解决CMC问题后很快获得批准。4月,礼来的IL-23单抗mirikizumab因生产制造问题收到拒绝信,但延期6个月后获批。再生元的8mg阿柏西普如是,产品在6月因第三方制造商问题收到CRL,经企业与CMO解决问题后,仅两月即获得FDA批准。但不是所有新药的CMC问题都能被轻易解决,尤其是生物药。生物药因分子量大、工艺复杂更易产生批间差异而出现CMC问题。11个拒批的新药几乎全部是生物药。而作为新兴疗法的CAR-T则更面临生产制造考验。近期,科济药业三款CAR-T产品CT053、CT041和CT071的临床试验被FDA按下暂停键,对其CMC合规问题存疑。对此肖申表示,FDA对CGT疗法审评的实践经验不长,以往无先例可循,因此对于新兴疗法FDA都要进行严格现场检查,且上市后的随访检查同样重要。例如已上市的CAR-T疗法Kymriah就在近期被FDA挑战,认为其生产制造存在新的重大问题,存在外来颗粒物污染和微生物污染。不论是CAR-T疗法在临床阶段或上市后出现生产制造问题,或是因CMC收到CRL延缓上市进程的新药,都提示着监管机构对CMC要求愈加严格,企业不可忽视CMC问题。综观2023年FDA拒绝和批准新药,患者需求是企业和监管机构绕不开的话题,新药研发是非常复杂且系统的工程,企业在临床研究过程中要积极和FDA保持沟通,及时获取FDA的审评意见并改进。同时,也启示着出海的中国企业,时刻关注FDA审评动向,制定满足海外患者需求的研发策略。下篇预告当肿瘤领域卷无可卷,制药巨头们还在拼什么?编辑 | 姚嘉yao.jia@PharmaDJ.com 总第2032期访问研发客网站可浏览更多文章www.PharmaDJ.com
临床3期基因疗法siRNA
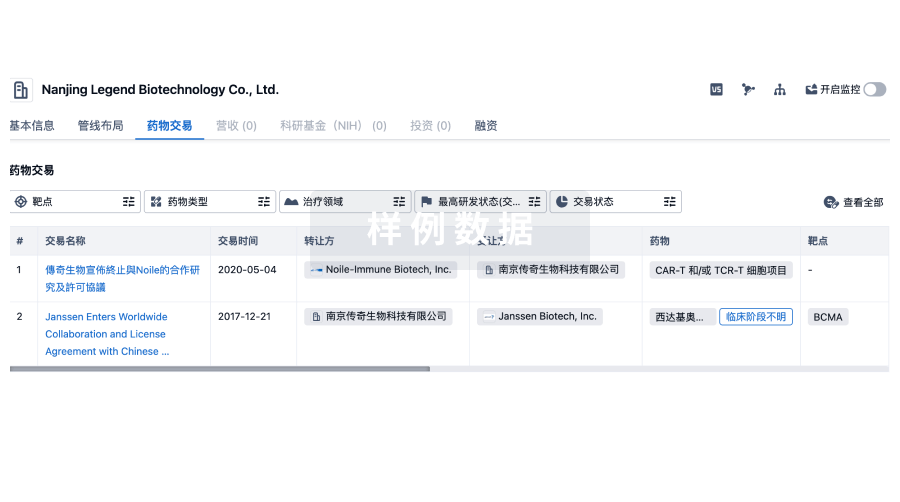
100 项与 Intercept Health 相关的药物交易
登录后查看更多信息
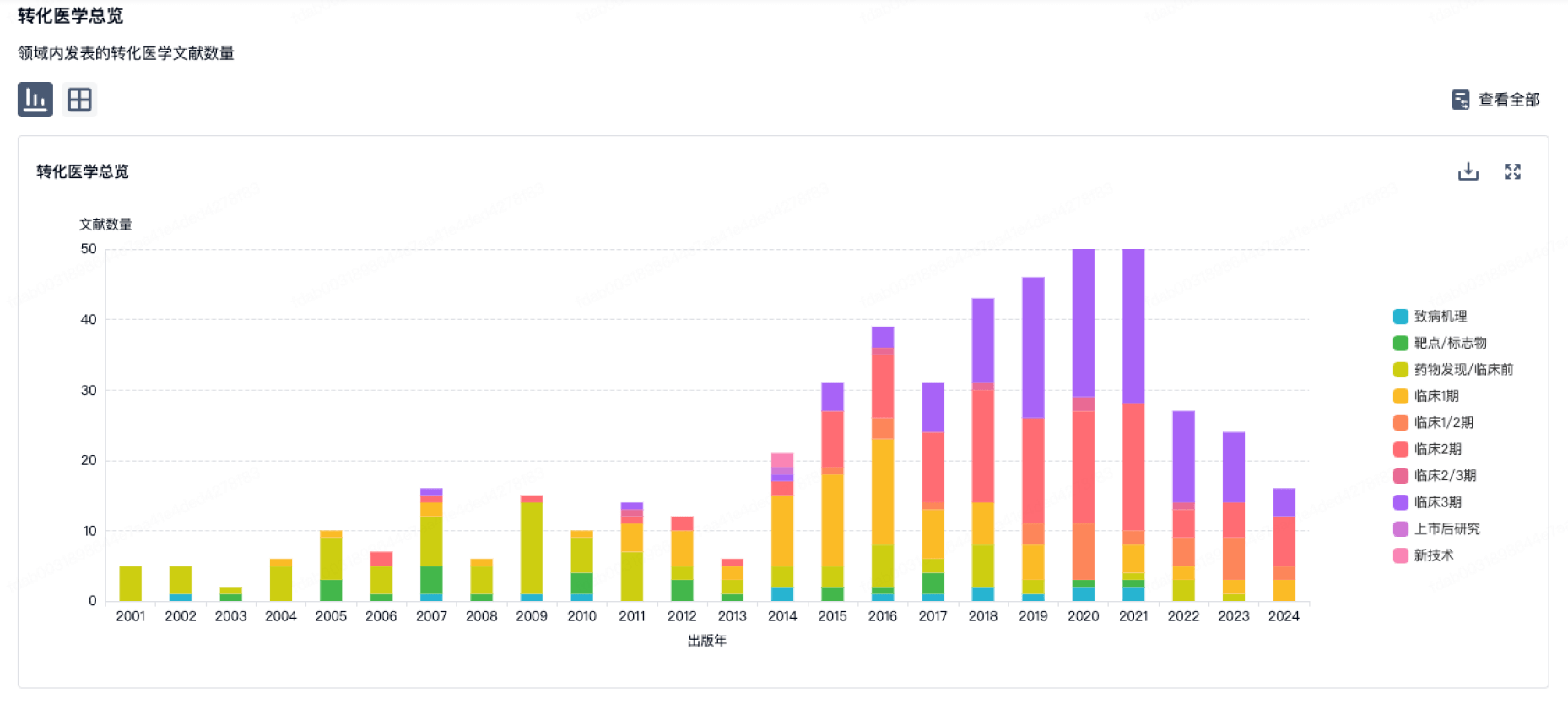
100 项与 Intercept Health 相关的转化医学
登录后查看更多信息
组织架构
使用我们的机构树数据加速您的研究。
登录
或

管线布局
2024年07月05日管线快照
无数据报导
登录后保持更新
药物交易
使用我们的药物交易数据加速您的研究。
登录
或
转化医学
使用我们的转化医学数据加速您的研究。
登录
或
营收
使用 Synapse 探索超过 36 万个组织的财务状况。
登录
或
科研基金(NIH)
访问超过 200 万项资助和基金信息,以提升您的研究之旅。
登录
或
投资
深入了解从初创企业到成熟企业的最新公司投资动态。
登录
或
融资
发掘融资趋势以验证和推进您的投资机会。
登录
或
标准版
¥16800
元/账号/年
新药情报库 | 省钱又好用!
立即使用
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用