预约演示
更新于:2025-05-07
Dysequilibrium Syndrome
失衡综合征
更新于:2025-05-07
基本信息
别名 Autosomal Recessive Cerebellar Ataxia with Mental Retardation、Autosomal Recessive Cerebellar Hypoplasia with Cerebral Gyral Simplification、CAMRQ1 + [22] |
简介- |
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

NCT06366230
Adding Urea to the Final Dialysis Fluid in Order to Prevent Dialysis Disequilibrium in Patients Who Need Aggressive Dialysis for Electrolyte Abnormalities

NCT06738446
Multicomponent Dietary Supplementation: Impact on Tear Secretion and Ocular Surface Inflammation in Dry Eye Syndrome Patients
NCT04948749
Drug Eluting Stenting and Aggressive Medical Treatment for Preventing Recurrent Stroke in Intracranial Atherosclerotic Disease Trial: a Prospective, Randomized, Open-labelled, Blinded End-point Trial (DREAM-PRIDE)
100 项与 失衡综合征 相关的临床结果
登录后查看更多信息
100 项与 失衡综合征 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2025-05-01European Journal of Ophthalmology
Torpedo maculopathy in a patient with DeSanto-Shinawi syndrome
Article
作者: Valente, Paola ; Federici, Matteo ; Catena, Gino ; Buzzonetti, Luca ; Petroni, Sergio ; Zinzanella, Gaetano ; Iarossi, Giancarlo ; De Sanctis, Carlo Maria
2025-04-01Circulation: Genomic and Precision Medicine
Natural History, Phenotype Spectrum, and Clinical Outcomes of Desmin (
DES
)-Associated Cardiomyopathy
Review
作者: Muller, Steven A. ; Rieder, Marina ; Leung, Doris G. ; Calkins, Hugh ; te Riele, Anneline S.J.M. ; Asatryan, Babken ; Carrick, Richard T. ; Zimmerman, Stefan L. ; Joseph, Emily ; Tichnell, Crystal ; Murray, Brittney ; Gasperetti, Alessio ; Barth, Andreas S. ; James, Cynthia A.
2025-03-01Journal of the Society for Cardiovascular Angiography & Interventions
Comparison of Different PCI Strategies for Coronary DES In-stent Restenosis: A Bayesian Network Meta-analysis
Review
作者: Khan, Rafay ; Shtembari, Jurgen ; Pant, Kailash ; Shrestha, Bishesh ; Poudel, Shraddha ; Katz, Daniel H ; Ali, Furkhan ; Dawadi, Sagun ; Oli, Prakash Raj ; Shrestha, Dhan Bahadur ; Mattumpuram, Jishanth
分析
对领域进行一次全面的分析。
登录
或
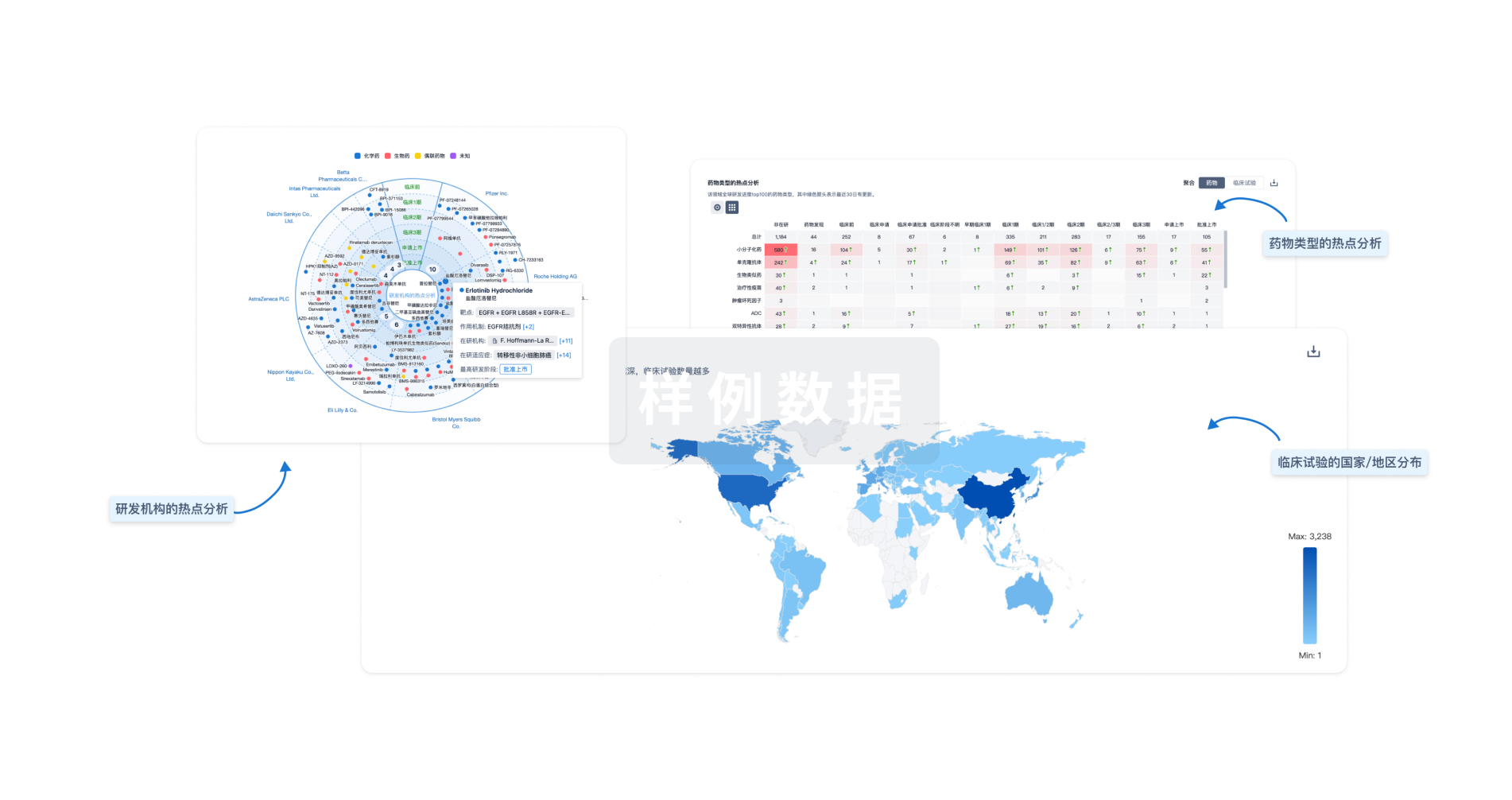
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用