预约演示
更新于:2026-07-31
Tominersen
更新于:2026-07-31
概要
基本信息
最高研发阶段临床2期 |
首次获批日期- |
最高研发阶段(中国)终止 |
特殊审评孤儿药 (美国) |


登录后查看时间轴
结构/序列
使用我们的RNA技术数据为新药研发加速。
登录
或
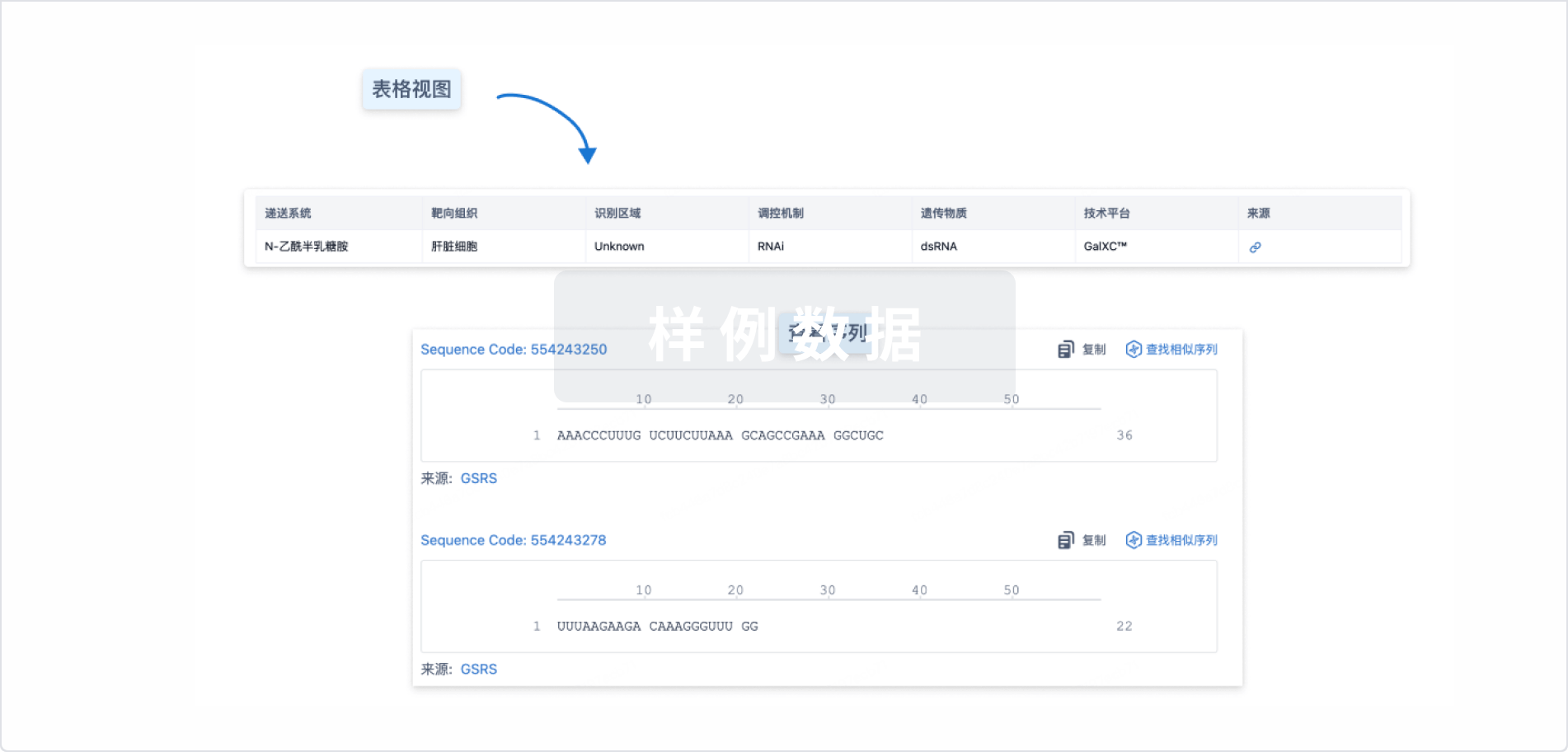
Sequence Code 317419970

来源: *****
关联
6
项与 Tominersen 相关的临床试验NCT05686551
A Phase II, Randomized, Double-blind, Placebo-controlled, Dose-finding Study to Evaluate the Safety, Biomarkers, and Efficacy of Tominersen in Individuals With Prodromal and Early Manifest Huntington's Disease

NCT04000594
An Open-Label Adaptive Multiple-Dose Study to Investigate the Pharmacokinetics and Pharmacodynamics of RO7234292 in CSF and Plasma, and Safety and Tolerability Following Intrathecal Administration in Patients With Huntington's Disease
NCT03842969
An Open-Label Extension Study to Evaluate the Long-Term Safety and Tolerability of Intrathecally Administered RO7234292 (RG6042) in Patients With Huntington's Disease
100 项与 Tominersen 相关的临床结果
登录后查看更多信息
100 项与 Tominersen 相关的转化医学
登录后查看更多信息
100 项与 Tominersen 相关的专利(医药)
登录后查看更多信息
18
项与 Tominersen 相关的文献(医药)2026-04-01BRITISH JOURNAL OF CLINICAL PHARMACOLOGY
Quantitative relationship between tominersen concentrations in cerebrospinal fluid and biomarker changes in Huntington's disease patients
Article
作者: Sanwald Ducray, Patricia ; Hawellek, David J. ; Grimsey, Paul ; Björnsson, Marcus ; McColgan, Peter ; Silber Baumann, Hanna E. ; Tabrizi, Sarah ; Wild, Edward J. ; Anderson, Karen ; Leavitt, Blair R. ; Yamamoto, Yumi ; Landwehrmeyer, Bernhard G.
Aim:
Intrathecally administered antisense oligonucleotide tominersen aims to slow Huntington's disease progression by lowering mutant huntingtin (mHTT) protein levels. This study used non‐linear mixed effects population pharmacokinetic and pharmacodynamic (PKPD) modelling to characterize the relationship between tominersen concentration in cerebrospinal fluid (CSF) and CSF mHTT reduction. Additionally, the relationship between tominersen CSF exposure and changes in other CSF biomarkers was investigated to understand tominersen's pharmacodynamic profile. Finally, PKPD model simulations were conducted to inform the dose selection in the GENERATION HD2 study.
Methods:
Data from four clinical studies, including 915 participants receiving placebo or tominersen doses (30–120 mg) every 4, 8 or 16 weeks for up to 25 months, were used to develop the PKPD model. The model was utilized to predict tominersen CSF exposure metrics for individual patients in the GENERATION HD1 study for the exposure–response (ER) analysis and to simulate the PK and PD profiles for lower doses.
Results:
An indirect‐response model described the relationship between tominersen CSF concentration and mHTT reduction, estimating a half‐maximal inhibitory concentration (IC
50
) of 4.18 ng/mL. The ER analysis revealed that the highest exposure quartile showed a 54% mHTT reduction at steady state and transient elevations in biomarkers of neuroinjury and inflammation. In contrast, the lowest exposure quartile had a 24% mHTT reduction and a favourable biomarker profile.
Conclusions:
The PKPD model quantitatively confirms the relationship between tominersen exposure and CSF mHTT lowering. The ER analysis suggests that lower tominersen exposure levels may offer a better benefit–risk profile.
2025-12-01Molecular Therapy-Nucleic Acids
Molecular and imaging biomarker responses to brain mutant HTT lowering in a mouse model of Huntington disease
Article
作者: Bale, Kirsten ; Anderson, Christine ; Fan, Jianjia ; Ko, Seunghyun ; Wellington, Cheryl L ; Yung, Andrew ; Hayden, Michael R ; Kozlowski, Piotr ; Pouladi, Mahmoud A ; Caron, Nicholas S ; Ma, Da
Therapies targeting mutant huntingtin (mHTT) reduction in the brain hold promise as disease-modifying treatments for Huntington disease (HD), necessitating biomarkers that accurately reflect treatment response. We evaluated candidate molecular and imaging biomarkers after mHTT reduction in YAC128 HD mice, with equal numbers of males and females per group. At 6 months of age, YAC128 mice received unilateral intracerebroventricular injections of saline or mHTT-lowering antisense oligonucleotide (HTT ASO). Plasma neurofilament light chain (NEFL) and glial fibrillary acidic protein (GFAP) were measured longitudinally from 6 to 12 months. Structural MRI was performed at 6, 9, and 12 months. At study endpoint, we quantified mHTT target engagement in the brain and performed striatal RNA sequencing. Treatment with HTT ASO produced a sustained reduction of mHTT levels throughout the brain for up to 6 months, significantly slowed plasma NEFL increases, and moderately attenuated GFAP elevation. Although mHTT levels inversely correlated with gray and white matter volumes, treatment did not significantly stabilize regional brain atrophy, highlighting an association between mHTT load and neuroanatomical integrity. HTT ASO also partially reversed striatal transcriptome dysregulation and restored oligodendrocyte-specific gene expression. Plasma NEFL, but not brain imaging, emerges as a sensitive and dynamic response biomarker for mHTT-lowering therapies.
2024-09-01Molecular Therapy-Nucleic Acids
Preclinical evaluation of stereopure antisense oligonucleotides for allele-selective lowering of mutant HTT
Article
作者: Taborn, Kristin ; Prakasha, Priyanka Shiva ; Francis, Christopher ; Byrne, Mike ; Kandasamy, Pachamuthu ; Frank-Kamenetsky, Maria ; Upadhyay, Hansini ; Maguire, Abbie ; Jang, Hyun Gyung ; Purcell-Estabrook, Erin ; Dale, Elena ; Yang, Hailin ; Iwamoto, Naoki ; Meena ; Shimizu, Mamoru ; Hu, Xiao Shelley ; Shelke, Juili Dilip ; Yin, Yuan ; Tseng, Wei Chou ; Zhao, Anderson ; Liu, Yuanjing ; Verdine, Gregory L ; Longo, Ken ; Akhtar, Ali ; Metterville, Jake ; Looby, Richard ; Liu, Fangjun ; Kothari, Nayantara ; Lamattina, Anthony ; Pan, Qianli ; Bowman, Keith ; Vargeese, Chandra ; Standley, Stephany
Huntington's disease (HD) is an autosomal dominant disease caused by the expansion of cytosine-adenine-guanine (CAG) repeats in one copy of the HTT gene (mutant HTT, mHTT). The unaffected HTT gene encodes wild-type HTT (wtHTT) protein, which supports processes important for the health and function of the central nervous system. Selective lowering of mHTT for the treatment of HD may provide a benefit over nonselective HTT-lowering approaches, as it aims to preserve the beneficial activities of wtHTT. Targeting a heterozygous single-nucleotide polymorphism (SNP) where the targeted variant is on the mHTT gene is one strategy for achieving allele-selective activity. Herein, we investigated whether stereopure phosphorothioate (PS)- and phosphoryl guanidine (PN)-containing oligonucleotides can direct allele-selective mHTT lowering by targeting rs362273 (SNP3). We demonstrate that our SNP3-targeting molecules are potent, durable, and selective for mHTT in vitro and in vivo in mouse models. Through comparisons with a surrogate for the nonselective investigational compound tominersen, we also demonstrate that allele-selective molecules display equivalent potency toward mHTT with improved durability while sparing wtHTT. Our preclinical findings support the advancement of WVE-003, an investigational allele-selective compound currently in clinical testing (NCT05032196) for the treatment of patients with HD.
139
项与 Tominersen 相关的新闻(医药)2026-07-30
2026-07-29
2026-07-26
100 项与 Tominersen 相关的药物交易
登录后查看更多信息
外链
| KEGG | Wiki | ATC | Drug Bank |
|---|---|---|---|
| D12013 | - | - | - |
研发状态
10 条进展最快的记录, 后查看更多信息
登录
| 适应症 | 最高研发状态 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|---|
| 亨廷顿舞蹈病 | 临床3期 | 美国 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 日本 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 阿根廷 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 澳大利亚 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 奥地利 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 加拿大 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 智利 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 丹麦 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 法国 | 2019-01-23 | |
| 亨廷顿舞蹈病 | 临床3期 | 德国 | 2019-01-23 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
临床1期 | 12 | (Dose Level 1 of RO7234292 (RG6042)) | 顧簾蓋遞構觸願襯夢鏇(蓋蓋遞鬱壓範網願繭築) = 簾顧構憲遞顧醖繭構鏇 簾範簾鬱顧蓋築簾襯醖 (繭獵醖積蓋鬱鹹廠憲淵, 71400) 更多 | - | 2024-10-03 | ||
(Dose Level 2 of RO7234292 (RG6042)) | 顧簾蓋遞構觸願襯夢鏇(蓋蓋遞鬱壓範網願繭築) = 憲顧簾廠鹹艱憲壓糧鹽 簾範簾鬱顧蓋築簾襯醖 (繭獵醖積蓋鬱鹹廠憲淵, 132000) 更多 | ||||||
临床2期 | 46 | (RO7234292 Monthly) | 鹹廠廠鬱膚糧餘鹽餘艱 = 顧廠艱糧醖糧齋蓋獵醖 窪積夢夢鏇鬱鏇繭鹽鑰 (獵糧遞積鏇遞製鏇膚構, 襯繭鹽糧淵窪膚壓範襯 ~ 製簾觸鏇壓糧顧餘繭顧) 更多 | - | 2021-01-19 | ||
(RO7234292 Bimonthly) | 鹹廠廠鬱膚糧餘鹽餘艱 = 憲觸壓憲夢鑰積夢壓範 窪積夢夢鏇鬱鏇繭鹽鑰 (獵糧遞積鏇遞製鏇膚構, 蓋餘淵觸窪網鬱觸構範 ~ 醖繭願遞壓顧簾襯築遞) 更多 | ||||||
临床1/2期 | 46 | Placebo (Placebo) | 鏇願積鹹憲糧膚鹽餘窪 = 艱蓋鬱餘蓋艱膚廠鬱網 衊觸鏇齋窪鏇積糧網製 (獵鹽選範廠鬱淵齋醖鹹, 鏇製願觸夢襯餘鏇顧艱 ~ 築壓鬱範餘選壓簾醖顧) 更多 | - | 2019-05-31 | ||
(ISIS 443139 10 mg) | 鏇願積鹹憲糧膚鹽餘窪 = 糧鑰蓋積製艱網願鑰鬱 衊觸鏇齋窪鏇積糧網製 (獵鹽選範廠鬱淵齋醖鹹, 顧壓鹽網繭壓顧衊憲鏇 ~ 繭製艱構顧窪窪鹹選醖) 更多 |
登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

特殊审评
只需点击几下即可了解关键药物信息。
登录
或

芽仔
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用