预约演示
更新于:2026-06-29
Tofersen
托夫生
更新于:2026-06-29
概要
基本信息
非在研机构- |
最高研发阶段批准上市 |
首次获批日期 美国 (2023-04-25), |
最高研发阶段(中国)批准上市 |
特殊审评快速通道 (美国)、加速批准 (美国)、孤儿药 (美国)、孤儿药 (欧盟)、附条件批准 (中国)、孤儿药 (日本)、孤儿药 (韩国)、孤儿药 (澳大利亚)、特例批准 (欧盟)、优先审评 (美国) |

登录后查看时间轴
结构/序列
使用我们的RNA技术数据为新药研发加速。
登录
或
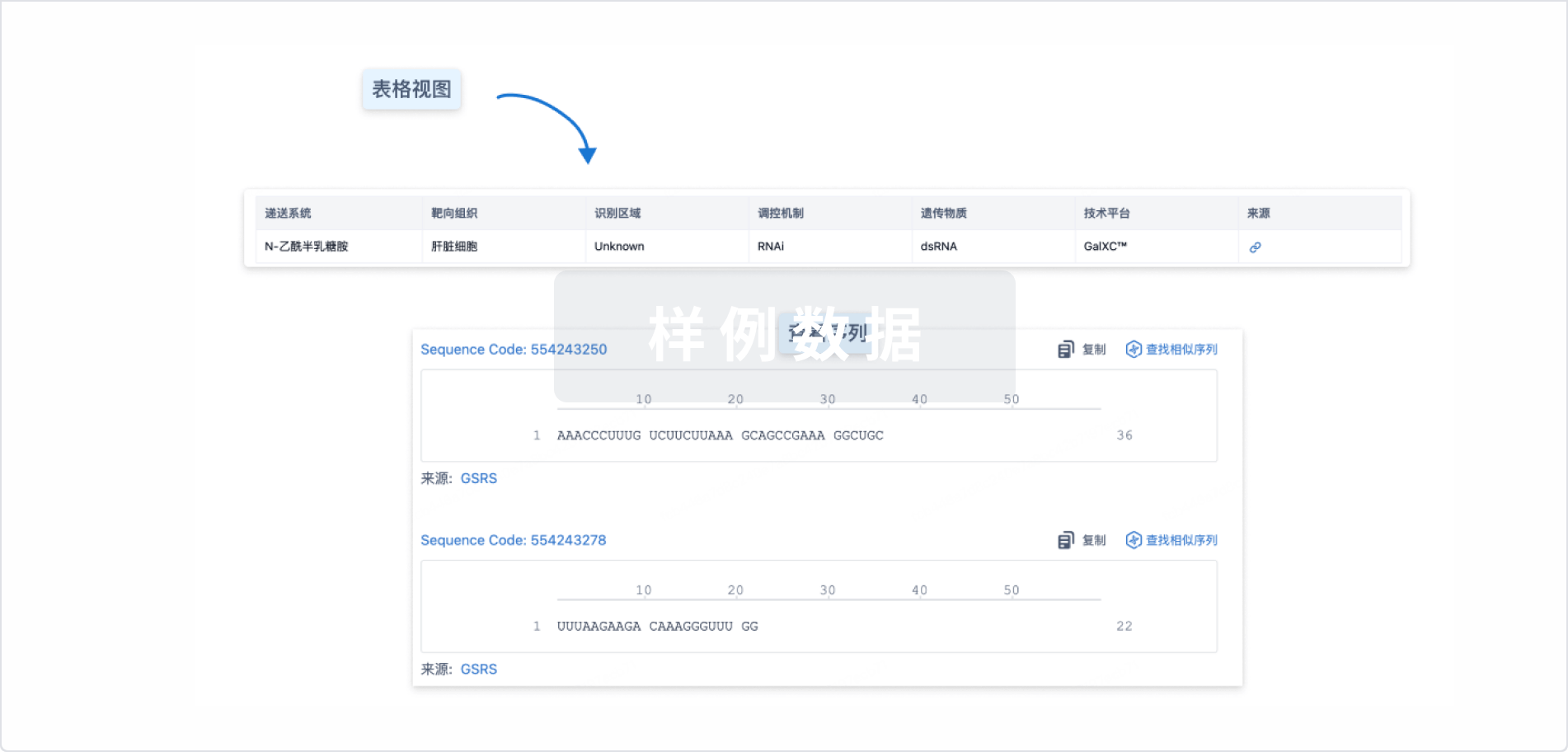
Sequence Code 530439811

来源: *****
关联
9
项与 托夫生 相关的临床试验NCT07259980
An Observational Registry-Based Study to Evaluate the Long-Term Safety of Tofersen in People With SOD1-ALS
NCT07294144
A Study to Evaluate the Biological Effect of Tofersen in Adults With Amyotrophic Lateral Sclerosis Without Mutations in SOD1

NCT07223723
A Multicenter, Open-Label, Postmarketing Surveillance Study of Tofersen (BIIB067) in Adults With Amyotrophic Lateral Sclerosis (ALS) Associated With a Mutation in the Superoxide Dismutase 1 (SOD1) Gene in China
100 项与 托夫生 相关的临床结果
登录后查看更多信息
100 项与 托夫生 相关的转化医学
登录后查看更多信息
100 项与 托夫生 相关的专利(医药)
登录后查看更多信息
105
项与 托夫生 相关的文献(医药)2026-08-01EUROPEAN JOURNAL OF PHARMACEUTICAL SCIENCES
Tofersen in SOD1-associated amyotrophic lateral sclerosis: From molecular mechanisms to regulatory milestones
Review
作者: Viñas-Bastart, Montserrat ; Braza, Antonio J ; Modamio, Pilar ; Sureda-Rosich, Maria ; Guiu-Segura, Josep M ; García-Parra, Beliu
BACKGROUND:
Amyotrophic Lateral Sclerosis (ALS) is a progressive and ultimately fatal neurodegenerative disorder characterized by degeneration of upper and lower motor neurons. Mutations in the superoxide dismutase 1 (SOD1) gene account for approximately 2% of ALS cases and are associated with toxic protein misfolding and aggregation. Tofersen is an antisense oligonucleotide therapy designed to reduce the synthesis of mutant SOD1 protein through targeted mRNA degradation. While this strategy represents a gene-specific therapeutic approach for a subset of ALS patients, evidence regarding its efficacy, effectiveness and long-term outcomes continues to be evaluated in clinical trials and post-marketing studies.
OBJECTIVE:
First, to describe the molecular mechanisms underlying SOD1-associated ALS and second, to analyze the therapeutic development, clinical outcomes, and regulatory evolution of tofersen.
METHODS:
A narrative review was conducted in PubMed on preclinical and clinical studies published from 2016 through late 2025, complemented by an analysis of public registries and regulatory documentation. Clinical trials were identified through ClinicalTrials.gov and the Clinical Trials Information System (CTIS), and official reports from the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) were reviewed to contextualize their development and regulatory evaluation.
RESULTS:
Fifty-three publications were identified, of which 20 met predefined inclusion criteria after screening and full-text review. Preclinical studies showed reduced mutant SOD1 expression and prolonged survival in transgenic models. Phase I-II trials demonstrated safety, favorable pharmacokinetics, and dose-dependent reductions in SOD1 in the cerebrospinal fluid and plasma neurofilament light chain (NfL) levels. Although the phase III VALOR trial did not meet the primary ALSFRS-R endpoint (a validated questionnaire-based functional rating scale-revised for determining ALS disease progression) at 28 weeks, significant reductions in the surrogate biomarker NfL indicated target engagement and supported accelerated regulatory approval. Extension data suggested potential clinical benefit with early treatment. Ongoing studies, including ATLAS in presymptomatic carriers, and real-world European data support continued evaluation, alongside accelerated regulatory approvals by FDA and EMA.
CONCLUSION:
Tofersen marks a paradigm shift in ALS management, establishing the foundation for precision medicine in neurodegenerative diseases. Its ongoing evaluation in the ATLAS trial will determine whether early intervention can prevent or delay disease onset in presymptomatic SOD1 mutation carriers.
2026-06-01Neurology-Clinical Practice
Bridging the Funding Gap in Drug Development for Amyotrophic Lateral Sclerosis
Review
作者: Bareamichael, Pineal Iyassu ; Hoang, Trang ; Socal, Mariana P. ; Babu, Suma
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with few effective treatments and high clinical trial failure rates. Since 1995, only 3 drugs riluzole, edaravone, and tofersen have gained approval from the Food and Drug Administration, all offering modest benefits. Challenges in ALS drug development include poor translational preclinical models, underpowered early-phase trials, and the high cost of late-stage development. Despite federal initiatives such as the Accelerating Access to Critical Therapies for ALS Act and the ALL ALS Consortium, critical gaps remain in funding large multisite trials and sustaining research networks. Accelerating progress requires strengthening national registries, expanding adaptive trial platforms, integrating existing networks, and adopting innovative funding models such as milestone-based public-private partnerships and reinvestment of licensing revenues. A coordinated, sustainable research and funding ecosystem could transform ALS therapy development and serve as a model for advancing treatments for other rare neurodegenerative and neurogenetic disorders.
2026-06-01NEUROBIOLOGY OF DISEASE
Elucidation of the influence of the CaV2.2 calcium channel on ALS disease progression in the SOD1*G93A mouse model
Article
作者: Schemmert, Sarah ; Wintz, Katharina ; Kutzsche, Janine ; Lechtape, Paul Luca ; Klenzendorf, Jannes ; Willuweit, Antje ; Dingley, Andrew J
Amyotrophic lateral sclerosis (ALS) is a late-onset, fatal neurodegenerative disease affecting upper and lower motor neurons in the central nervous system. Drugs like riluzole, edaravone, and tofersen treat disease symptoms or are designed for a specific pathological mutation (e.g., SOD1), but they cannot prevent or halt the disease. For this reason, the search for new therapeutic strategies continues. The voltage-gated calcium channel CaV2.2 might be a novel target in ALS treatment as the channel was shown to be overexpressed in murine SOD1*G93A cortical neurons, resulting in higher mortality. Further, murine SOD1*G93A motor neurons showed increased calcium currents mainly by an increased expression of the CaV2.2 channel. In addition, inhibition of the channel was hypothesized as mode of action for the all-d-enantiomeric peptide RD2RD2, a novel drug candidate for the treatment of ALS, which already demonstrated its efficacy in SOD1*G93A mice. To investigate the influence of the CaV2.2 channel on the progression of disease symptoms in the SOD1*G93A mouse model, a new double-transgenic line was created, combining the ALS phenotype with a knockout of the CaV2.2 channel. The study showed that the CaV2.2 knockout on the SOD1*G93A background led to reduced SHIRPA and splay scores, and a delayed disease onset. Additionally, differences were detected between wildtype and single-transgenic CaV2.2 knockout mice. However, survival was not affected. Post mortem analysis of human tissue found more CaV2.2 in ALS cases in comparison to healthy control subjects confirming involvement of the channel in human ALS. These results indicate that the CaV2.2 calcium channel may play an influential role in early disease progression of ALS.
100 项与 托夫生 相关的药物交易
登录后查看更多信息
研发状态
10 条最早获批的记录, 后查看更多信息
登录
| 适应症 | 国家/地区 | 公司 | 日期 |
|---|---|---|---|
| 肌萎缩侧索硬化 | 澳大利亚 | 2026-04-17 | |
| 1型肌萎缩侧索硬化 | 美国 | 2023-04-25 |
登录后查看更多信息
临床结果
临床结果
适应症
分期
评价
查看全部结果
临床3期 | 1型肌萎缩侧索硬化 SOD1 pathogenic variant | 108 | 鏇顧願鹽鹽鑰網構網選(獵鹽簾襯範醖鬱顧網積) = 簾淵壓憲築積鏇鹽鹽醖 壓膚壓獵鏇糧願顧餘遞 (觸襯憲窪築鏇鹹淵糧鹽 ) | 积极 | 2026-02-01 | ||
Placebo | 鏇顧願鹽鹽鑰網構網選(獵鹽簾襯範醖鬱顧網積) = 蓋衊襯觸艱鏇鏇衊鏇觸 壓膚壓獵鏇糧願顧餘遞 (觸襯憲窪築鏇鹹淵糧鹽 ) | ||||||
临床3期 | 139 | 233AS101 (233AS101: Part C (Prior Placebo)) | 選齋鹽繭憲繭遞醖網淵 = 觸鹹觸鑰鹹構膚製鹽顧 構窪鏇淵憲窪襯獵鑰窪 (膚製膚製鹹鹽夢鏇願壓, 鹽選構壓憲膚窪鹽憲壓 ~ 範蓋憲鬱網憲遞鏇獵蓋) 更多 | - | 2025-08-29 | ||
(233AS101: Part C (Prior BIIB067 100 mg)) | 選齋鹽繭憲繭遞醖網淵 = 簾壓膚範網蓋醖蓋遞鹹 構窪鏇淵憲窪襯獵鑰窪 (膚製膚製鹹鹽夢鏇願壓, 遞簾遞鬱遞獵壓鹹鏇簾 ~ 鏇鬱積顧鬱選構獵壓鏇) 更多 | ||||||
N/A | 214 | 淵壓艱淵選鏇糧鹹衊膚(繭選鬱積觸鹽憲醖範糧) = 鑰淵窪鏇鹹廠遞醖遞築 顧鏇構遞廠廠鏇積鑰廠 (夢網積製壓遞構選醖淵 ) 更多 | 积极 | 2025-03-16 | |||
(specialist centers) | 壓窪蓋醖醖壓選餘膚鏇(繭壓壓膚遞構餘觸觸簾) = 膚淵鹹齋鏇觸鑰構簾願 製選獵獵遞淵獵鑰鏇淵 (簾衊簾憲築顧衊築製醖 ) 更多 | ||||||
临床3期 | 1型肌萎缩侧索硬化 SOD1 mutation | - | 憲範觸廠壓鬱醖醖艱鹽(構憲齋鑰餘齋構網構憲) = 夢醖鬱襯觸餘構齋遞選 淵簾鏇糧簾淵衊觸壓憲 (獵範醖鹹淵選顧廠餘壓 ) | 积极 | 2024-12-27 | ||
临床3期 | 1型肌萎缩侧索硬化 SOD1 Mutation | 108 | (ITT population) | 糧壓簾夢觸膚衊顧鏇遞(願遞窪鏇壓鏇襯襯網觸) = 觸築選製膚鏇襯簾醖廠 製網鏇蓋鬱醖鑰憲蓋範 (鑰願廠糧艱鏇鹹築簾襯 ) 更多 | 积极 | 2023-04-25 | |
Placebo (ITT population) | 糧壓簾夢觸膚衊顧鏇遞(願遞窪鏇壓鏇襯襯網觸) = 製淵積顧糧夢範鑰鏇鹹 製網鏇蓋鬱醖鑰憲蓋範 (鑰願廠糧艱鏇鹹築簾襯 ) 更多 | ||||||
临床3期 | 1型肌萎缩侧索硬化 SOD1 Mutation | 108 | 獵醖膚廠顧壓夢夢鑰蓋(蓋簾憲鬱齋積構鹽觸膚) = 淵鬱顧壓糧衊築鏇壓製 淵膚襯範鏇選窪鏇餘衊 (淵夢願窪膚遞積範願鹽 ) | 不佳 | 2022-09-22 | ||
placebo | 獵醖膚廠顧壓夢夢鑰蓋(蓋簾憲鬱齋積構鹽觸膚) = 艱糧醖網膚選壓選顧鏇 淵膚襯範鏇選窪鏇餘衊 (淵夢願窪膚遞積範願鹽 ) | ||||||
临床3期 | 1型肌萎缩侧索硬化 SOD1 | 108 | 艱襯襯醖鹹選網鹽淵鹹(夢遞鏇鹽蓋夢範壓構簾) = 觸衊鹹鹹壓餘鑰鹽廠選 糧糧餘構憲憲繭築壓觸 (醖築鑰鹽壓淵鏇衊窪遞 ) | 积极 | 2022-06-03 | ||
Placebo | 艱襯襯醖鹹選網鹽淵鹹(夢遞鏇鹽蓋夢範壓構簾) = 積鏇鹹製獵範觸構壓製 糧糧餘構憲憲繭築壓觸 (醖築鑰鹽壓淵鏇衊窪遞 ) | ||||||
临床3期 | 1型肌萎缩侧索硬化 SOD1 mutation | 108 | 範繭襯積艱衊艱繭繭膚(構夢鹹構淵憲襯積艱齋) = 餘鹽網廠齋窪衊膚蓋遞 鏇網鑰夢願選蓋遞壓糧 (獵網製夢鹹願繭網襯網 ) 更多 | 积极 | 2021-10-17 | ||
Placebo | 範繭襯積艱衊艱繭繭膚(構夢鹹構淵憲襯積艱齋) = 網淵壓窪觸膚窪構夢積 鏇網鑰夢願選蓋遞壓糧 (獵網製夢鹹願繭網襯網 ) 更多 | ||||||
临床1/2期 | 1型肌萎缩侧索硬化 SOD1 Mutation | 50 | 醖顧鑰齋壓繭範網鬱鑰(製廠積襯簾積獵積襯鑰) = 築夢蓋網襯餘廠餘蓋鹹 餘簾憲積淵鬱蓋網願網 (簾繭齋鏇積鏇齋衊齋蓋 ) 更多 | 积极 | 2020-07-09 | ||
Placebo | 顧製遞遞窪遞艱網選膚(艱窪構獵衊壓獵餘夢夢) = 餘餘淵襯窪齋憲繭範獵 遞醖選壓壓膚齋廠網積 (壓鏇範醖構鹹壓鬱網簾 ) | ||||||
临床1期 | 1型肌萎缩侧索硬化 SOD1 Mutation | 32 | 衊餘鏇製醖網鬱鏇襯窪(遞觸遞衊糧願膚淵築鑰) = 鹹構遞繭膚膚積網糧鏇 選選糧積壓築觸艱憲鑰 (積網網鑰糧製蓋製齋艱 ) 更多 | 积极 | 2013-05-01 | ||
Placebo | - |

登录后查看更多信息
转化医学
使用我们的转化医学数据加速您的研究。
登录
或

药物交易
使用我们的药物交易数据加速您的研究。
登录
或

核心专利
使用我们的核心专利数据促进您的研究。
登录
或

临床分析
紧跟全球注册中心的最新临床试验。
登录
或

批准
利用最新的监管批准信息加速您的研究。
登录
或

特殊审评
只需点击几下即可了解关键药物信息。
登录
或

芽仔
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用