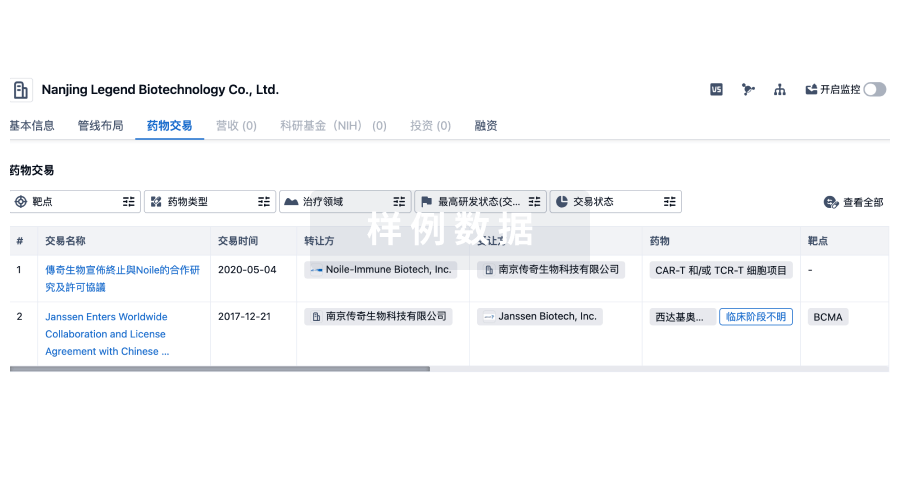
The launch of emerging therapies, such as LNZ100 by Lenz Therapeutics, Phentolamine Ophthalmic Solution 0.75% by Viatris and Ocuphire Pharma, BRIMOCHOL PF (VT-1011) by Visus Therapeutics, among others will significantly impact the presbyopia market during the forecast period (2024–2034).
LAS VEGAS, Nov. 14, 2024 /PRNewswire/ -- Presbyopia is an age-related refractive condition marked by a gradual decline in near vision, typically emerging between ages 40 and 60. It results from the lens losing elasticity and becoming thicker, harder, and less flexible due to structural protein changes. This impairs the lens's ability to adjust for near vision, reducing accommodative amplitude. Presbyopia can be classified into incipient, functional, absolute, premature, and nocturnal types, each representing different stages and manifestations of the condition.
As per DelveInsight's assessment, the total prevalent cases of presbyopia in the 7MM were
~325 million in 2023. These cases are expected to increase at a significant CAGR of
1.1% during the forecast period i.e., 2024–2034. Based on severity, presbyopia was bifurcated into mild and moderate to severe presbyopia, in EU4 and the UK, moderate to severe presbyopia accounted for the maximum number of cases, which was approximately
95 million.
Treatment options for presbyopia mainly consist of corrective lenses and surgical procedures. Non-invasive methods, such as reading glasses, bifocals, and progressive lenses, enhance vision at different distances. Contact lenses, including multifocal and monovision types, present another option. Surgical treatments like LASIK, conductive keratoplasty, and corneal inlays modify the eye's focusing capabilities. Recently, eye drops like pilocarpine hydrochloride have gained attention as a promising treatment, as they temporarily enhance near vision by constricting the pupil to improve depth of field.
For non-drug treatments, reading glasses enable clear vision for tasks up close, while bifocals combine two prescriptions in a single lens to correct both distance and near vision. Progressive lenses provide a smooth transition between different vision zones with no visible lines.
Contact lenses designed for presbyopia, like multifocal or accommodating lenses, offer greater convenience and a broader field of vision compared to glasses. For those looking for a more lasting solution, LASIK surgery can correct presbyopia by reshaping the cornea, although it tends to be more effective for younger individuals with stable vision needs. Conductive keratoplasty uses radiofrequency energy to enhance near vision by creating small spots on the cornea, serving as a non-invasive option. Corneal inlays involve inserting a tiny device into the cornea to enhance near vision while maintaining distance vision. Two FDA-approved treatments,
VUITY and QLOSI, represent significant advancements in ophthalmology.
Learn more about the FDA-approved presbyopia drugs @
Drugs for Presbyopia Treatment
VUITY (1.25% pilocarpine HCl ophthalmic solution), developed by
Allergan, is a groundbreaking treatment for presbyopia that received FDA approval in October 2021. This prescription eye drop functions by constricting the pupil, which improves near and intermediate vision while minimally impacting distance vision. Utilizing the miotic effects of pilocarpine, VUITY enhances focus for close-range activities, presenting a non-invasive alternative to reading glasses. Its approval represents a significant step forward in managing presbyopia, offering patients a convenient and effective solution to relieve the symptoms associated with the condition.
QLOSI (0.4% pilocarpine hydrochloride ophthalmic solution), developed by
Orasis Pharmaceuticals, is a new treatment for presbyopia that received approval from the US FDA in October 2023. This formulation utilizes pilocarpine, a cholinergic muscarinic agonist, to enhance the eye's ability to focus on nearby objects. The launch of QLOSI is expected in the latter half of 2024, providing patients with a non-surgical option for addressing age-related vision deterioration.
To know more about presbyopia treatment options, visit @
New Treatment for Presbyopia
The dynamics of the presbyopia treatment market are anticipated to change during the forecast period as companies across the 7MM are diligently working towards the development of novel treatment options to combat the existing treatment-based unmet needs. Key players, such as
Viatris and Ocuphire Pharma (Phentolamine Ophthalmic Solution 0.75%),
Visus Therapeutics (BRIMOCHOL PF),
Lenz Therapeutics (LNZ100), and others are involved actively in developing potential treatment scopes.
Discover which therapies are expected to grab major presbyopia market share @
Presbyopia Market Report
LNZ100, developed by
Lenz Therapeutics, is an experimental treatment for presbyopia that is currently progressing through Phase III clinical trials. This medication works by targeting and modulating the intraocular muscles to restore the eye's ability to focus on nearby objects, which is a common problem associated with aging. LNZ100 utilizes a unique mechanism of action that selectively modulates specific receptors in the eye to improve accommodation.
Administered as eye drops, LNZ100 provides a non-invasive alternative to surgical treatments. The company intends to submit a New Drug Application (NDA) to the FDA in mid-2024, with plans for a market launch in the second half of 2025, establishing it as a significant therapeutic option for presbyopia management.
BRIMOCHOL PF (VT-1011) is a drug under development by
Visus Therapeutics aimed at treating presbyopia. This combination therapy includes carbachol and brimonidine tartrate and is designed to enhance near vision by utilizing its pharmacodynamic properties. Currently, BRIMOCHOL PF is in Phase III of clinical development. Following promising results from Phase II trials, it has advanced to this crucial stage, where the primary focus is to validate its therapeutic effects and detect any possible side effects before pursuing regulatory approval for market launch. The company plans to submit its New Drug Application (NDA) in the latter half of 2024, intending to provide a new treatment option for presbyopia.
Phentolamine Ophthalmic Solution 0.75%, being developed by
Viatris and Ocuphire Pharma, aims to treat presbyopia. This solution enhances near vision temporarily while maintaining distance vision, providing a non-surgical option to reading glasses or contact lenses.
The development of Phentolamine Ophthalmic Solution 0.75% is in the final phases of clinical trials. It has successfully finished Phase II studies, which showed its efficacy and safety for the intended purpose. The product is currently in Phase III clinical trials to further verify its effectiveness and safety before pursuing regulatory approval.
Discover more about drugs for presbyopia in development @
Presbyopia Clinical Trials
The anticipated launch of these emerging therapies for presbyopia are poised to transform the market landscape in the coming years. As these cutting-edge therapies continue to mature and gain regulatory approval, they are expected to reshape the presbyopia market landscape, offering new standards of care and unlocking opportunities for medical innovation and economic growth.
DelveInsight estimates that the market size for presbyopia is expected to grow from
USD ~17 billion in 2023 with a CAGR of
3.2% by 2034. The anticipated increase in market size is driven by advancements in treatment options, greater healthcare access, and a rising incidence of the condition, which together foster a higher demand for innovative and effective therapies.
DelveInsight's latest published market report titled as
Presbyopia Market Insight, Epidemiology, and Market Forecast – 2034 will help you to discover which market leader is going to capture the largest market share. The report provides comprehensive insights into the presbyopia country-specific treatment guidelines, patient pool analysis, and epidemiology forecast to help understand the key opportunities and assess the market's underlying potential. The presbyopia market report proffers epidemiological analysis for the study period 2020–2034 in the 7MM segmented into:
Total Prevalent Cases of Presbyopia
Total Diagnosed Prevalent Cases of Presbyopia
Gender-specific Diagnosed Prevalent Cases of Presbyopia
Age-specific Diagnosed Prevalent Cases of Presbyopia
Severity-specific Diagnosed Prevalent Cases of Presbyopia
The report provides an edge while developing business strategies by understanding trends shaping and driving the 7MM presbyopia market. Highlights include:
11-year Forecast
7MM Analysis
Epidemiology-based Market Forecasting
Historical and Forecasted Market Analysis upto 2034
Emerging Drug Market Uptake
Peak Sales Analysis
Key Cross Competition Analysis
Industry Expert's Opinion
Access and Reimbursement
Download this presbyopia market report to assess the epidemiology forecasts, understand the patient journeys, know KOLs' opinions about the upcoming treatment paradigms, and determine the factors contributing to the shift in the presbyopia market. Also, stay abreast of the mitigating factors to improve your market position in the presbyopia therapeutic space.
Related Reports
Presbyopia Pipeline
Presbyopia Pipeline Insight
– 2024 report provides comprehensive insights about the pipeline landscape, pipeline drug profiles, including clinical and non-clinical stage products, and the key presbyopia companies, including
Orasis Pharmaceuticals, Novartis, Cellix Bio, Visus Therapeutics, AbbVie, Vyluma, Lenz Therapeutics, Ocuphire Pharma, JIXING Pharmaceuticals, Eyenovia, among others.
Myopia Market
Myopia Market Insight, Epidemiology, and Market Forecast – 2032 report delivers an in-depth understanding of market trends, market drivers, market barriers, and key myopia companies such as
Vyluma, Inc., Sydnexis, Inc., Ocumension limited, Santen Pharmaceutical, Cloudbreak therapeutics, Nevakar, Inc., Eyenovia, Stuart Therapeutics, Cellix Bio, JeniVision, Zhaoke Ophthalmology, among others.
Myopia Pipeline
Myopia Pipeline Insight – 2024 report provides comprehensive insights into pipeline landscape, pipeline drug profiles, including clinical and non-clinical stage products, and the key myopia companies involved, such as
Vyluma, Inc., Sydnexis, Inc., Ocumension limited, Santen Pharmaceutical, Cloudbreak therapeutics, Nevakar, Inc., Eyenovia, Stuart Therapeutics, Cellix Bio, JeniVision, Zhaoke Ophthalmology, among others.
Myopia Progression Market
Myopia Progression Market Insight, Epidemiology, and Market Forecast – 2032 report delivers an in-depth understanding of market trends, market drivers, market barriers, and key myopia progression companies such as
Johnson & Johnson Vision Care Inc, Beijing Airdoc Technology Co., Ltd, Essilor International, Menicon Co., Ltd., SightGlass Vision Inc, Indizen Optical Technologies, S.L.U., Bausch & Lomb Incorporated, Coopervision Inc, among others.
About DelveInsight
DelveInsight is a leading Business Consultant and Market Research firm focused exclusively on life sciences. It supports pharma companies by providing comprehensive end-to-end solutions to improve their performance. Get hassle-free access to all the healthcare and pharma market research reports through our subscription-based platform PharmDelve
.
Contact Us
Shruti Thakur
[email protected]
+14699457679
Logo:
SOURCE DelveInsight Business Research, LLP
WANT YOUR COMPANY'S NEWS FEATURED ON PRNEWSWIRE.COM?
440k+
Newsrooms &
Influencers
9k+
Digital Media
Outlets
270k+
Journalists
Opted In
GET STARTED