预约演示
更新于:2025-05-07
Ischaemic cerebral infarction
缺血性脑梗死
更新于:2025-05-07
基本信息
别名 Ischaemic cerebral infarction、Ischemic cerebral infarction、ischemic cerebral infarction + [3] |
简介- |
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
DRKS00035601
Cerebral perfusion measurement using transcranial ultrasound.A comparative study between contrast-enhanced ultrasound (CEUS) and CT perfusion (CT-P): a prospective, monocentric, exploratory clinical study.
NCT06786988
Cerebral Safety After Pulsed-Field Ablation of Atrial Fibrillation

IRCT20211206053309N2
The Impact of Telenursing on Caregiver Burden in homecare Providers of Patients with ischemic stroke
100 项与 缺血性脑梗死 相关的临床结果
登录后查看更多信息
100 项与 缺血性脑梗死 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2025-12-01Human & Experimental Toxicology
Rhynchophylline promotes microglia phenotypic transformation and repair of cerebral ischaemic injury through the JAK2/STAT3 pathway
Article
作者: Ju, Meng ; Bai, Peng ; Li, Caixia ; Li, Yao ; Yin, Luwei ; Wang, Laicang
2025-07-01Experimental Neurology
Identification of NMT1/MA/VPS15 signal pathway as potential therapeutic target in rat cerebral ischemia injury
Article
作者: Chen, Juan ; Wan, Qi ; Bao, Shuguang ; Bu, He ; Shen, Na ; Liu, Rui ; Wang, Yajie ; Li, Zhuo ; Wang, Hui ; Dong, Shan ; Lin, Tao ; Ma, Wenlong
2025-06-01Journal of Controlled Release
Brain-targeting biomimetic nanozyme enhances neuroprotection in ischemic stroke by remodeling the neurovascular unit
Article
作者: Luan, Yuxia ; Jiang, Yue ; Tang, Mingtan ; Shan, Lingling ; Wang, Luyao ; Peng, Tingting ; He, Wenxiu ; Wan, Bo ; Song, Yan
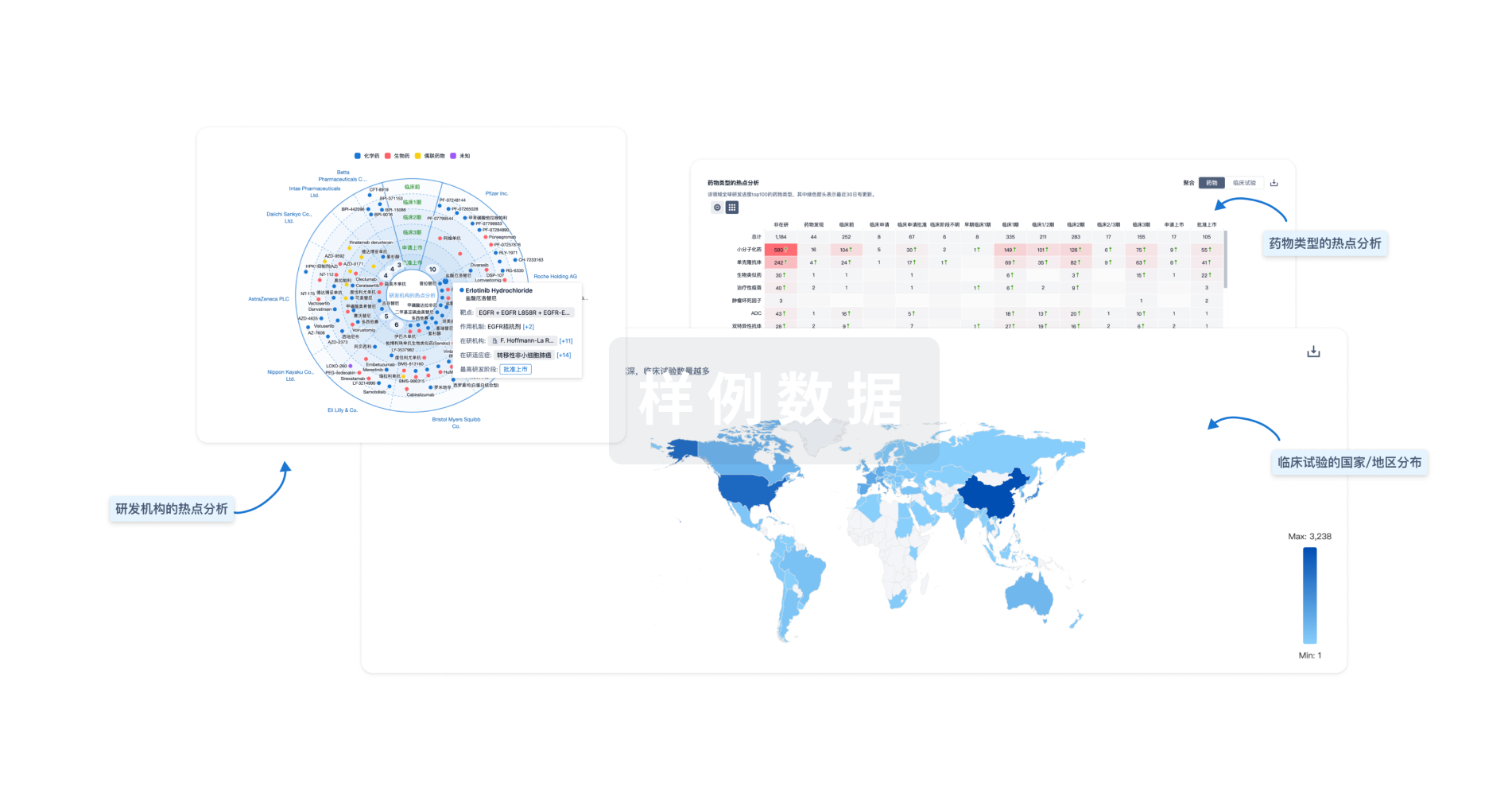
分析
对领域进行一次全面的分析。
登录
或
芽仔
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用