预约演示
更新于:2025-05-07
Xeroderma Pigmentosum
色素性干皮症
更新于:2025-05-07
基本信息
别名 Angioma Pigmentosum Atrophicum、Angioma pigmentosum atrophicum、Atrophoderma Pigmentosum + [37] |
简介 A rare, pigmentary, and atrophic autosomal recessive disease. It is manifested as an extreme photosensitivity to ULTRAVIOLET RAYS as the result of a deficiency in the enzyme that permits excisional repair of ultraviolet-damaged DNA. |
关联
靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |

靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |


靶点 |
作用机制 |
在研机构 |
原研机构 |
在研适应症 |
非在研适应症 |
最高研发阶段 |
首次获批国家/地区 |
首次获批日期 |
NCT06330324
Reproductive Options in Inherited Skin Diseases: an International Observational Cohort Study
NCT05484570
Natural History Study for DNA Repair Disorders
JPRN-jRCT2051210181
A multicenter, double-blind, placebo-controlled, two-group crossover study and long-term open study evaluating the efficacy and safety of NPC-15 in patients with xeroderma pigmentosum (XP) sunburn enhancement. - XP-1
100 项与 色素性干皮症 相关的临床结果
登录后查看更多信息
100 项与 色素性干皮症 相关的转化医学
登录后查看更多信息
登录后查看更多信息
2025-05-01Human Genetics
The molecular landscape of hereditary ataxia: a single-center study
Article
作者: Baldan, Federica ; Lonigro, Incoronata Renata ; Verriello, Lorenzo ; Allegri, Lorenzo ; Zucco, Jessica ; Dal Secco, Chiara ; Betto, Elena ; Damante, Giuseppe ; Faletra, Flavio ; Bregant, Elisa ; Mio, Catia
2025-05-01DNA Repair
Identification of a novel pathogenic XPC:c.2420 + 1 G>C variant in a patient with xeroderma pigmentosum
Article
作者: Faatih, Mukhlissul ; Ulhaq, Zulvikar Syambani ; Ratnangganajati, Estu
2025-04-01BMJ Case Reports
Xeroderma pigmentosum type C with prominent cutaneous manifestations and subclinical neuroimaging abnormalities
Article
作者: Saadeh, Imad ; Alhamood, Mohammed Mamdoh ; Nassar, Nassar ; Alsuleiman, Yassin
分析
对领域进行一次全面的分析。
登录
或
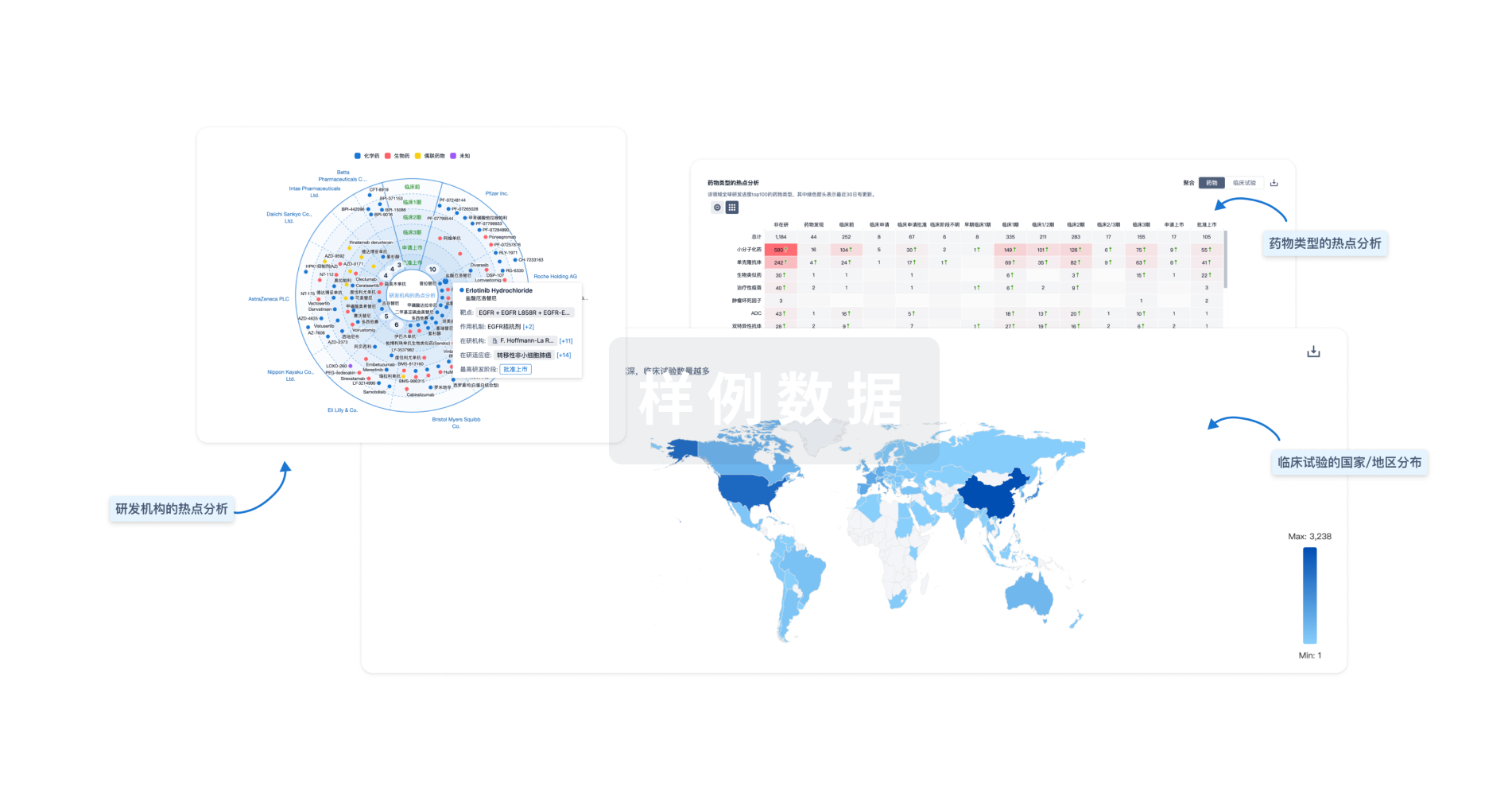
芽仔
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步
立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。
生物序列数据库
生物药研发创新
免费使用
化学结构数据库
小分子化药研发创新
免费使用