预约演示
新一代4-1BB激动剂的开发 :旨在更安全、有效
临床结果免疫疗法
在过去的十年中,用于癌症免疫治疗的4-1BB激动剂的临床开发引起了人们的极大兴趣。进入临床的第一代4-1BB激动型抗体Urelumab(BMS-663513)和Utomilumab(PF-05082566)分别因肝毒性或无效而失败。这两种抗体在亲和力、4-1BB表位识别以及决定FcγR交联活性的isotype上存在差异。基于这一经验,最近开发了一系列多样化的第二代4-1BB激动剂,旨在解决第一代激动剂的一些问题。目前许多第二代4-1BB激动剂药物也已进入了临床I期和II期研究。本文就第二代差异化的4-1BB激动剂分子做一个简要介绍。
第一代4-1BB激动剂
激动型4-1BB抗体的临床开发始于2005年的urelumab,这是一种人源化的抗4-1BB IgG4抗体。虽然最初的疗效还不错,但发生了两起由肝毒性引起的致命性不良事件。随后的研究表明,当urelumab在安全剂量(0.1mg/kg)下给药时,其效果非常有限。第二个4-1BB激动型抗体utomilumab是一种全人源抗4-1BB IgG2抗体,于2011年进入临床(NCT01307267)。与urelumab不同,utomilumab没有引起重大毒副作用,但它无论是作为单一疗法还是与利妥昔单抗联合使用疗效均有限,最终导致临床开发中断。
Urelumab和utomilumab表现出完全不同的特征,被认为可能影响毒性和疗效。Urelumab的亲和力(22或16.6 nM)高于utomilumab(69或71.2 nM)。然而,4-1BB激动性抗体的亲和力似乎不是激动性活性和肝毒性诱导的关键,而是由FC介导的交联和表位结合驱动的。
人IgG2(用于utomilumab)与人IgG4(用于urelumab)一样,表现出较低的铰链灵活性。因此,铰链灵活性可能不能解释urelumab较高的激动活性。Fc介导的交联,特别是与FcγRIIB的结合可以促进激动型抗体的活性。作为一种IgG4,urelumab对FcγRIIB具有更高的亲和力,因此可能介导更强的FcγRIIB依赖的激动性活性,但这可能不是utomilumab和 urelumab激动性差异的唯一因素。然而,FcγRIIB在肝脏中的交联与4-1BB激动型抗体诱导的肝炎有关。
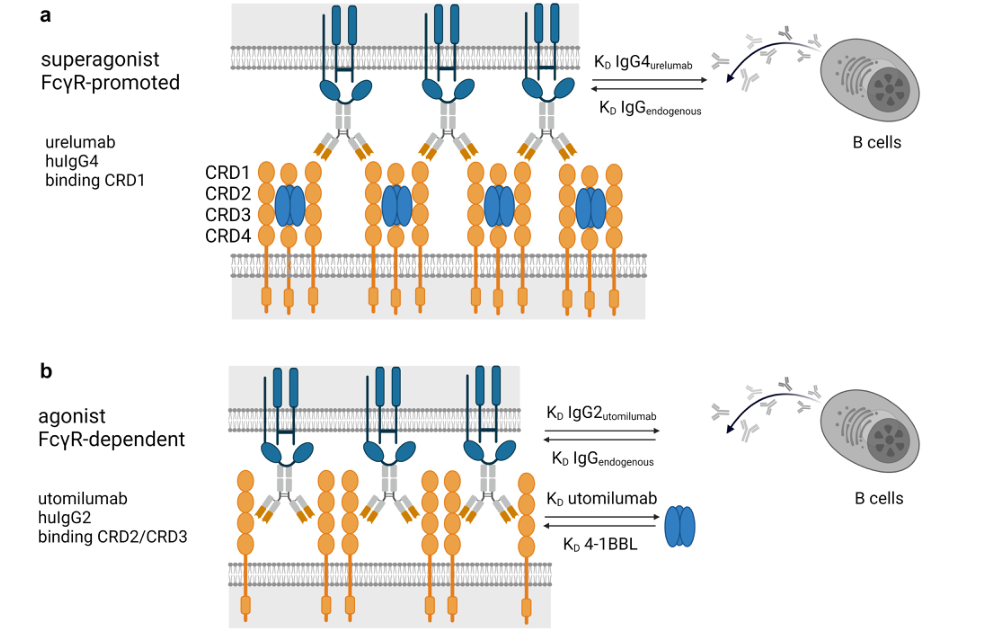
Urelumab和utomilumab识别的不同表位已被广泛研究,以了解它们对观察到的差异的影响。Urelumab与4-1BB膜远端富含半胱氨酸的假重复序列结构域(CRD1)的表位结合,其不干扰配体4-1BBL的结合。utomilumab与4-1BB结构域CRD2和CRD3结合,并与天然配体竞争。因此,内源性4-1BBL与utomilumab竞争4-1BB受体结合并限制其4-1BB受体聚集活性,而urelumab的活性被内源性4-1BBL增强,导致超级激动性特性(图1)。
Preview
来源: 生物制药小编
图1. Urelumab与utomilumab的区别
第二代4-1BB激动剂
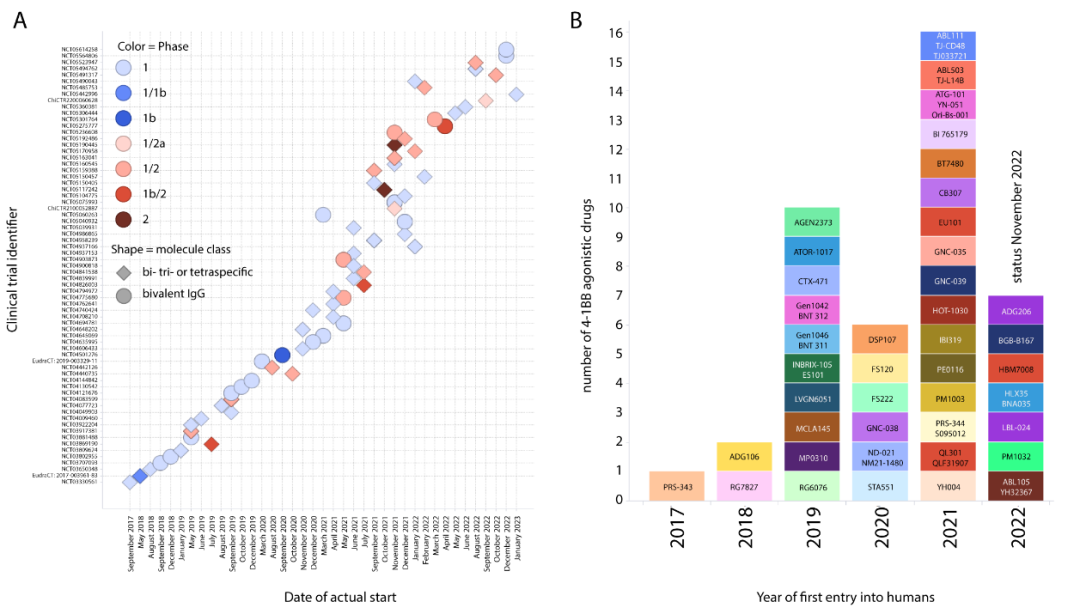
根据已发布的报告和数据,目前至少有40多种4-1BB激动剂药物进入了临床试验(图2)。
Preview
来源: 生物制药小编
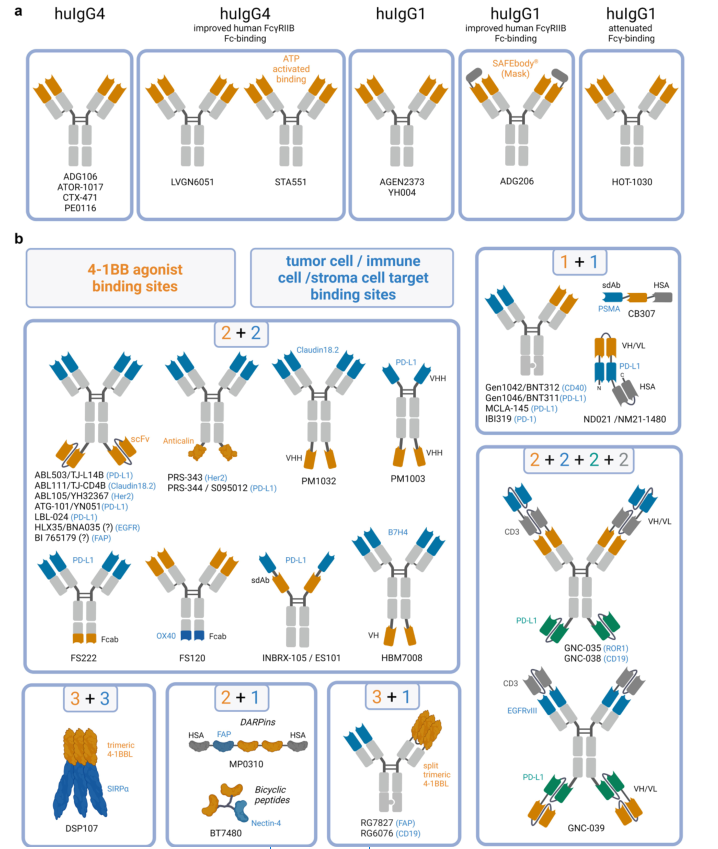
另有几十种新的4-1BB激动剂正在积极的临床前开发中,预计未来将有更多的4-1BB激动剂进入临床。图2. 进入临床阶段的4-1BB激动剂药物继urelumab和utomilumab之后进入临床第二代4-1BB激动剂具有不同的分子的分子设计,大致可以分为两大类:基于IgG的分子和双、三或四特异分子(图3)。
Preview
来源: 生物制药小编
图3. 第二代4-1BB激动剂的不同分子设计基于IgG的4-1BB激动剂
Preview
来源: 生物制药小编
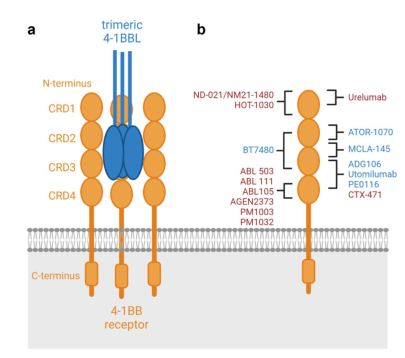
图4.第二代4-1BB激动剂结合表位示意图
双、三或四特异性4-1BB激动剂
另一组第二代4-1BB激动剂是双、三或四特异性4-1BB激动剂(图3b)。
基于IgG的双、三或四特异性的药物通常表现出Fc的修饰,以消除与FcγR的结合,但保持与FcRn的结合。消除与FcγR的结合的目的是防止全身活性,并抑制肝毒性。因此,这些类型的激动剂的原理是严格通过与交联靶部位的结合来提供交联,以提供更好的安全性。一些三特异性分子缺乏Fc(CB307,ND021/NM21-1480,MP0310),但通过依靠与HSA的结合来增加半衰期。
大多数分子仍然显示抗体骨架或包括抗体衍生结合域,例如scFv(ABL503/TJ-L14B,ABL111/TJ-CD4B,ABL105/YH32367,ATG-101/YN051,LBL-024), VH(CB307), VH/VL(ND021/NM21-1480),5sdAb(INBRIX-105/ES101)或VHH(PM1003,PM1032)。FAP-4-1BBL(RG7827)和CD19-4-1BBL(RG6076)是基于人4-1BBL胞外结构域融合到IgG1框架上的抗体融合蛋白;DSP107是一种没有抗体成分的三聚体融合蛋白。PRS-343和PRS-344/S095012在IgG4骨架的重链的C末端融合了两个anticalins。FS120和FS222含有一个二价结合Fcab(与抗原结合的Fc区),它整合在Fc区C端。其他新的分子包括MP0310,使用“设计的锚蛋白重复蛋白”(DARPins)作为抗体框架的替代品;BT7480,基于双环肽平台,产生具有抗体样亲和力和特异性的分子。
表位结合
据预测,4-1BB激动剂的大小和表位可能对最佳突触的形成起作用。例如,对于ND021/NM21-1480,靶向4-1BB N末端(膜远端) 表位比膜近端表位功能要好。有报道预测140 Å是最佳的突触间距,这可能使较小的分子具有优势。
不同4-1BB激动剂一个重要的特征是4-1BBL非阻断或阻断特性。一方面,高水平的可溶性4-1BBL可能会阻碍4-1BB激动剂的功能。另一方面,一些临床前数据显示,抑制4-1BBL反向信号可导致更好的T细胞激活。4-1BB激动剂与CRD1(如Urelumab,HOT1030)或CRD4(如ABL50,ABL111,65AGEN2373,51PM1003,PM1032,49CTX-47135)的至少双价结合而不被4-1BBL阻断可能导致全身性激活,特别是含有高水平的可溶性4-1BBL。然而,ND-021/NM21-1480只能与一个4-1BB受体结合,因此在没有同时与PD-L1结合的情况下,不能使4-1BB受体超交联化。在PD-L1存在下,可溶性4-1BBL水平也可以增强ND-021/NM21-1480的功能。
对于大多数4-1BB激动剂,表位还尚未披露,尽管它已被预测为确定安全性和功能性的主要参数之一。
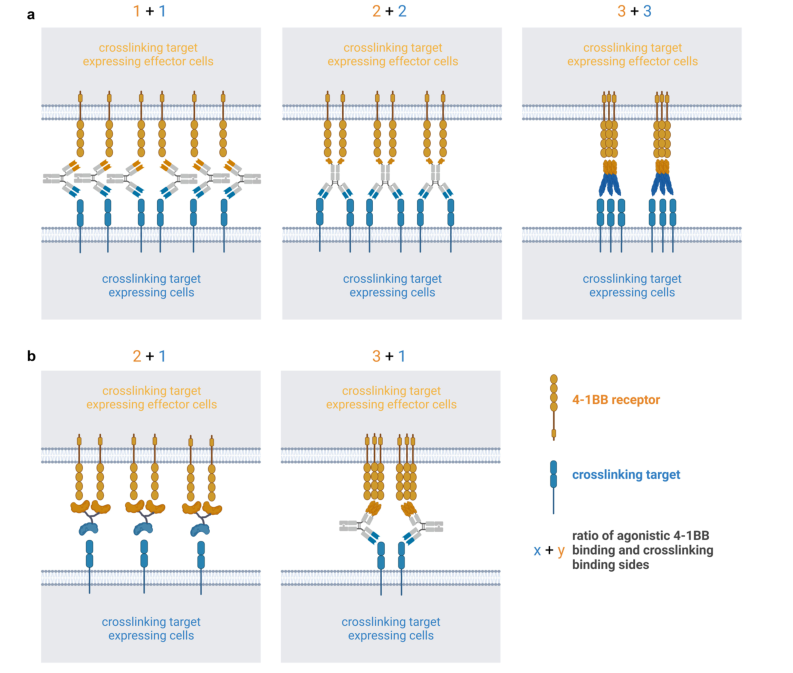
结合位点与结合亲和力之比
Preview
来源: 生物制药小编
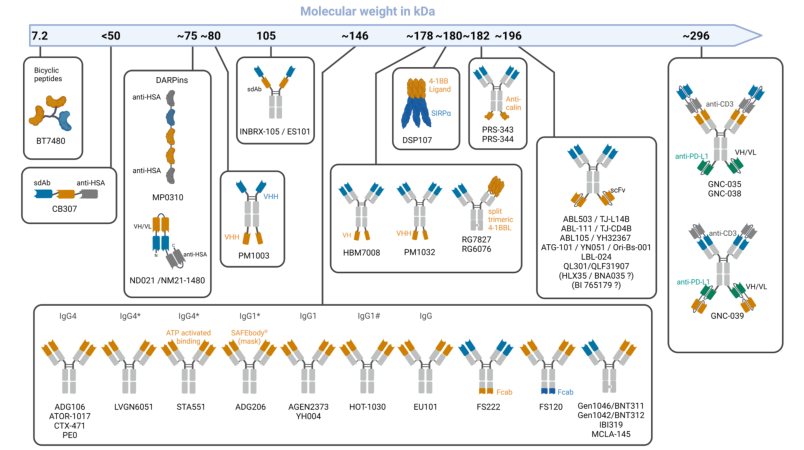
图5.不同4-1BB受体与交联/靶标结合位点比对超交联性的理论影响这些在分子质量较高的情况下尤其重要(图6),例如,在低交联靶表达的组织中的激活(如FAP)。虽然在临床前已经证明了高4-1BB/靶结合位点比的优点,但这在多大程度上转化为临床仍然未知。例如,在靶标表达高的情况下,具有较高的4-1BB/靶标结合位点比率的分子所提供的优势可能并不显著。此外,高的靶标结合亲和力导致靶结合位点和4-1BB结合位点之间差异具有诱导更好的功能性的潜在优势,特别是在低靶表达的情况下。
Preview
来源: 生物制药小编
图6. 4-1BB激动剂的相对分子质量
Preview
来源: 生物制药小编
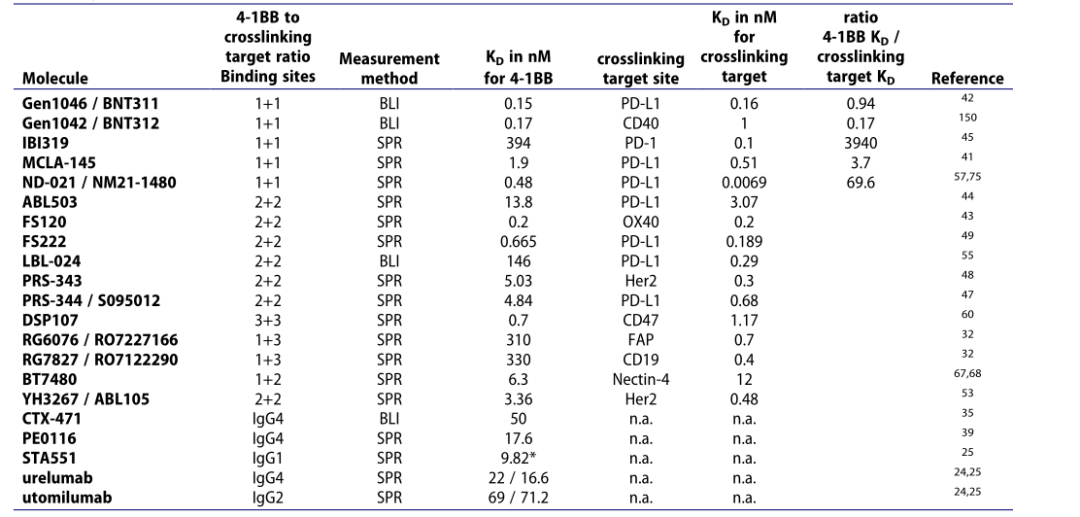
表1. 4-1BB激动剂药物结合 4-1BB KD和靶标kD
动物模型研究4-1BB激动剂的局限性
有研究表明,4-1BB抗体的结合表位和Fc Crosslinking而非高亲和力是降低肝毒性以及抗肿瘤活性的关键。但这些研究大多是在同基因小鼠或人类4-1BB转基因小鼠上进行的,存在一定的局限性,对人的临床可译性也有限。当4-1BB激动剂在小鼠中使用时,必须考虑到小鼠FcγR结合的跨物种反应性。Kamata-Sakurai和他的同事通过设计被称为StA-MB(用于STA551)和URE-MB(用于Urelumab)的小鼠替代物来显示小鼠IgG1(MB)的工程恒定区域来模拟小鼠的FcγRII交联,从而克服其中的一些限制。
此外,还必须考虑到,小鼠4-1BB/4-1BBL复合体是二聚体,不同于人4-1BB/4-1BBL三聚体,而且人4-1BB与小鼠4-1BBL不相互作用。因此,人4-1BB转基因小鼠模型不能用于预测内源性4-1BBL竞争或阻断作用。最近,人4-1BB/人4-1BBL双转基因小鼠也被开发,以提高其对人类的可译性。
受体占有率与最佳剂量寻找
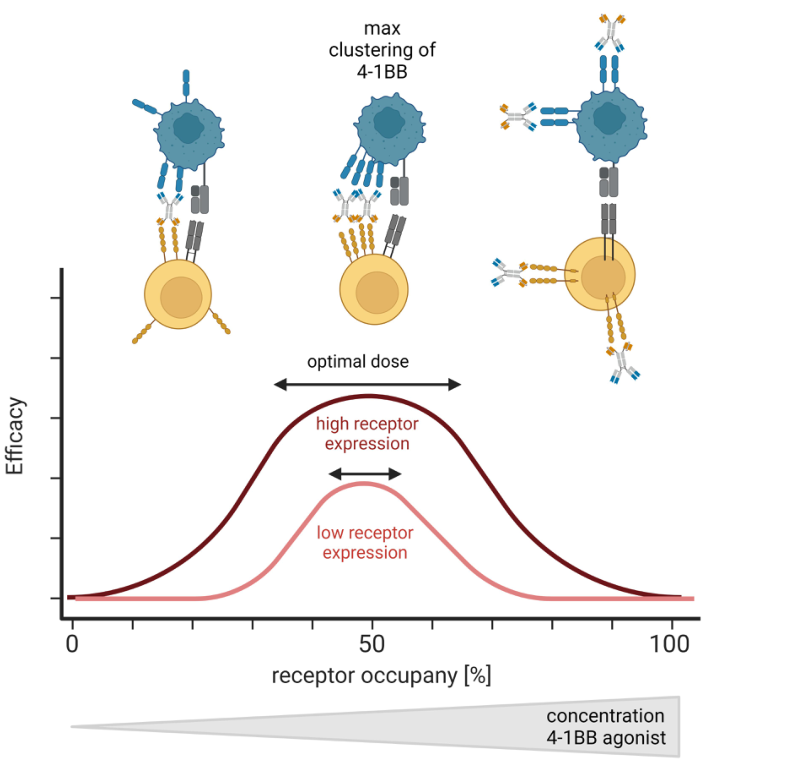
受体占有率已被预测为激动型抗体最佳剂量发现的关键因素。如图7所示,假设在受体占有率为~50%时的最大效应,导致由靶细胞、药物和表达4-1BB的效应细胞的三元复合体提供最佳4-1BB超聚集。较高的药物浓度导致受体占有率为100%但将阻止最佳三聚体的形成并导致钟形激活曲线。因此,了解4-1BB的表达是必要的,但这具有挑战性,因为4-1BB在T细胞上的表达受到严格控制。正常情况下,CD3激活的PBMC来源的T细胞表面具有4-1BB的表达,但5天后便检测不到4-1BB的表达。4-1BB的激活可将4-1BB的表达延长至第5天,并且CD8+T细胞上的4-1BB表达强于CD4+T细胞,导致肿瘤微环境中CD8+T细胞优先结合4-1BB激动剂。同时, 4-1BB激动剂也可能激活4-1BB内化、以及4-1BB脱落等。
Preview
来源: 生物制药小编
图7. 预测的最佳剂量取决于受体占有率,导致钟形活性曲线
最佳剂量的预测不仅取决于受体的最佳占有率,还取决于可溶性4-1BB、可溶性交联靶蛋白和可溶性4-1BBL介导的可能的外周下沉。机制效应也将发挥作用,如受体结合后的内化,4-1BB/靶的亲和力关系,或肿瘤组织穿透的因素,如分子大小和PK。因此,临床上寻找最佳剂量是具有挑战性的,特别是在具有不同4-1BB和交联靶表达的患者群体中。已报道临床试验的4-1BB激动剂的给药方案如图8所示。总体上,选择了固定剂量或基于体重的剂量(mg/kg)。正在测试不同的给药方案,包括Q1W、Q2W、Q3W、Q4W和Q6W给药。到目前为止,关于最佳剂量发现的数据有限。
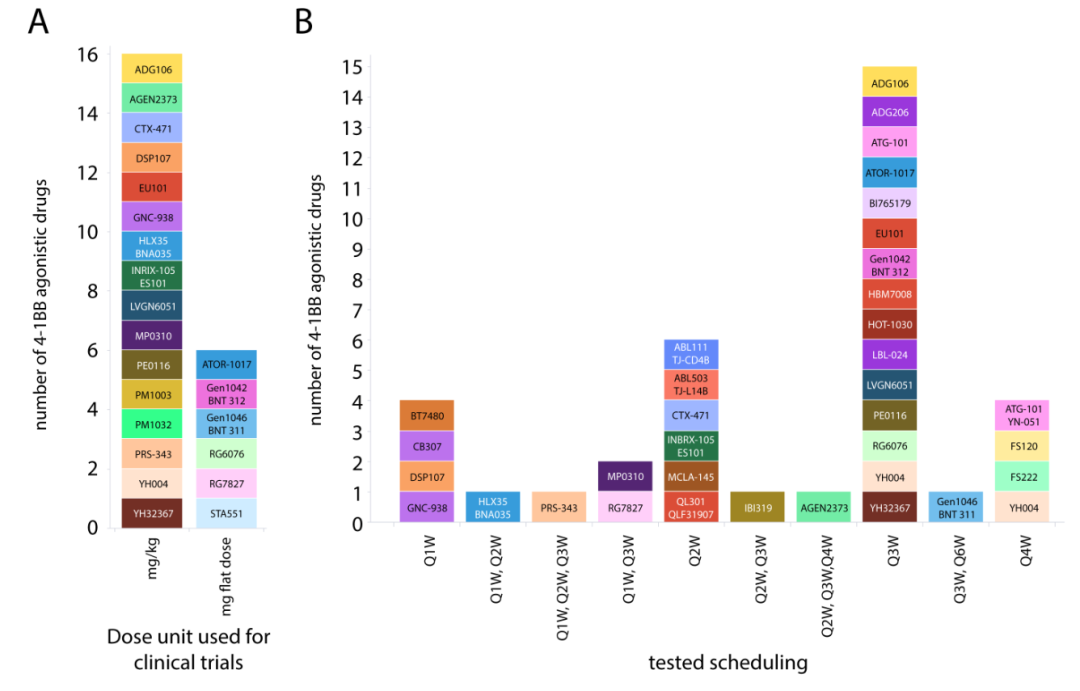
Preview
来源: 生物制药小编
图8. 已报道临床试验的4-1BB激动剂的给药方案
联合疗法
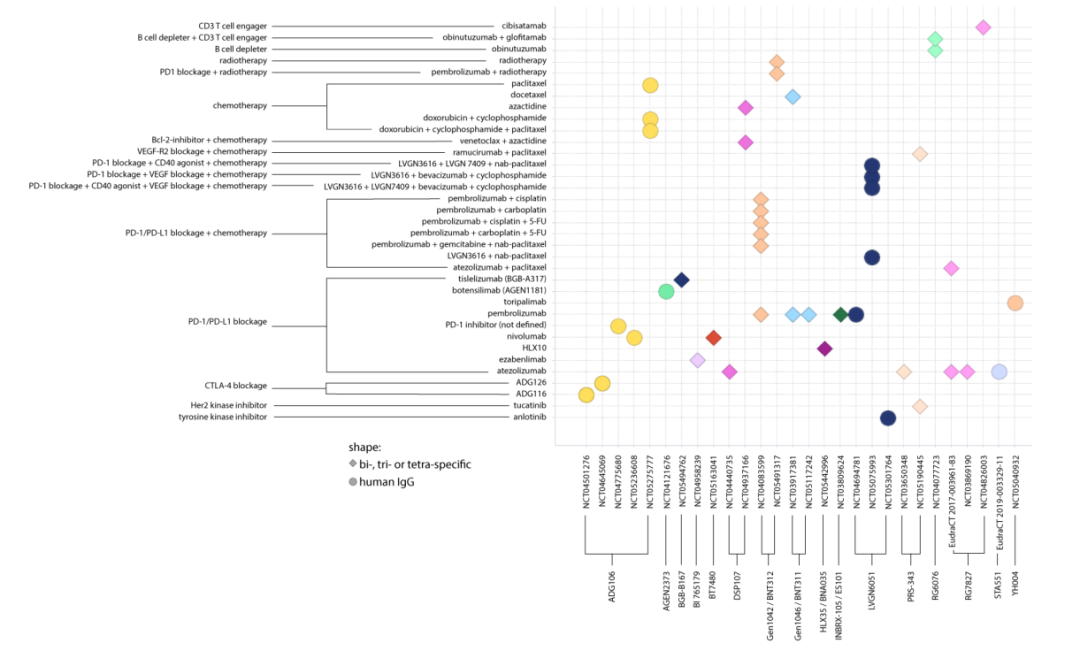
Preview
来源: 生物制药小编
在CD40和4-1BB双特异性抗体GEN1042/BNT312治疗期间,50例患者中有1例出现4级转氨酶升高,经皮质类固醇治疗后缓解。第二代4-1BB激动剂在实体肿瘤中的疾病控制率似乎约为56-70%。Stable disease率(SD)在为26-70%之间,总有效率(ORR)在3.6-40%之间。但这些数据主要是在正在进行的I期试验中获得的。到目前为止,所有已公布临床数据的第二代4-1BB激动剂总体上都有良好的安全性和耐受性。常见的不良反应多为1级和2级。一些不良反应,如肺炎、瘙痒、皮疹或注射相关不良反应、ALT/AST升高、甲状腺功能减退、腹泻和结肠炎,在癌症免疫治疗中很常见,可定义为常见的免疫相关不良反应(irAEs)。这些irAEs是否与更好或更差的结果相关尚不完全清楚,但一项研究表明,在癌症免疫治疗期间发生的irAEs与较好的结果相关。因此,在未来,irAEs的管理将是改进癌症免疫治疗的重要任务。
小结
在过去的5年里,对第二代4-1BB激动剂的研究有了很大的进展,正在实施几种策略来克服第一代4-1BB激动剂的缺陷。开发中的4-1BB激动剂分子高度多样化。虽然这些分子在大小(分子量)、半衰期、亲和力、交联靶标以及靶标结合位点的价态上不同,但它们都旨在实现安全和有效的4-1BB超聚集。到目前为止,所有4-1BB激动剂似乎都表现出良好的安全性和耐受性。但哪种分子设计是诱导抗肿瘤反应的理想分子,并具有良好的安全性,仍有待证实。无论设计如何,克服临床发展中的挑战,如寻找最佳剂量和最佳联合合策略,将是新一代4-1BB激动剂能够获得成功的关键。参考出处1.The emerging landscape of novel 4-1BB (CD137) agonistic drugs for cancer immunotherapy.2.Epitope and Fc-mediated Crosslinking, but not High Affinity, Are Critical for Antitumor Activity of CD137 Agonist Antibody with Reduced Liver Toxicity.
内容来源于网络,如有侵权,请联系删除。
机构
-药物
生物医药百科问答
全新生物医药AI Agent 覆盖科研全链路,让突破性发现快人一步



立即开始免费试用!
智慧芽新药情报库是智慧芽专为生命科学人士构建的基于AI的创新药情报平台,助您全方位提升您的研发与决策效率。
立即开始数据试用!
智慧芽新药库数据也通过智慧芽数据服务平台,以API或者数据包形式对外开放,助您更加充分利用智慧芽新药情报信息。